Worcester Polytechnic Institute, Department of Biology & Biotechnology
University of Massachusetts Medical School, Program in Molecular Medicine
RNA Therapeutics Institute
Howard Hughes Medical Institute

Description
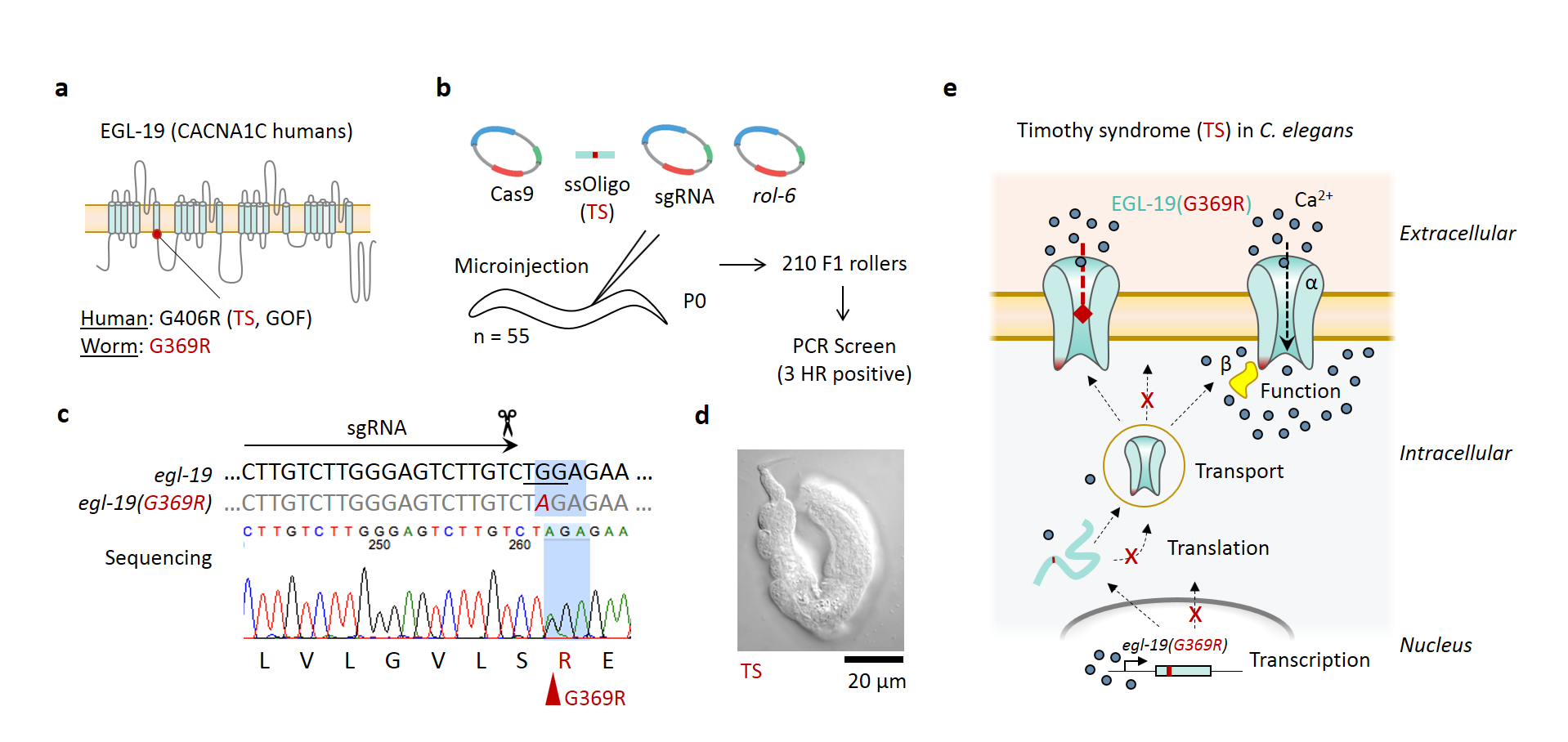
Timothy syndrome (TS) type 1 is a rare genetic human disease caused by a gain-of-function (GOF) missense mutation G406R in the L-type voltage-gated calcium channel (VGCC) a1 subunit gene CACNA1C, which results in severe cardiac arrhythmia and autism (Splawski et al. 2004). The C. elegans ortholog is EGL-19 (Fig. 1a), which is expressed in muscle and neurons (Lee et al. 1997). GOF mutations in egl-19 cause myotonic phenotypes, while reduction-of-function (ROF) mutants are flaccid, variably elongated, and egg-laying defective, and lethal mutations cause paralysis with arrested elongation at the two-fold stage (Pat) (Lee et al. 1997). Additionally, treatment of embryos with nemadipine-A, an antagonist of L-type VGCCs, causes a severe variable abnormal (Vab) phenotype like some ROF hatchlings (Kwok et al. 2006). Neural and muscle cells derived from patients with TS type 1 also yield impaired channel inactivation (Yazawa et al. 2011; Paşca et al. 2011). Therefore, we hypothesized that insertion of the human TS type 1 GOF mutation in the C. elegans genome (G369R) by CRISPR-Cas9 homologous recombination (HR) would cause observable changes in calcium dynamics and serve as a new animal disease model of TS to broadly investigate molecular mechanisms in vivo.
Instead, introduction of the TS type 1 human GOF mutation in C. elegans resulted in homozygous animals that closely resemble the Vab phenotype (Fig. 1b-d), similar to ROF mutant hatchlings and wild type embryos treated with the L-type VGCC antagonist nemadipine-A. This phenotype was observed in three independently edited lines, suggesting it resulted from the human TS mutation rather than a cis loss-of-function mutation in egl-19 generated by the CRISPR-Cas9 editing. Further, heterozygous animals appeared wild type, which is consistent with recessive inheritance of ROF and lethal mutations in egl-19. Taken together, this human mutation appears to dysregulate egl-19 function in C. elegans indirectly, such as through expression, trafficking, or channel kinetics at the cell membrane (Fig. 1e). Evaluation of gene expression, protein localization, and functional imaging or electrophysiology in this new animal model of TS type 1 are needed to distinguish among these possibilities.
This result demonstrates that CRISPR-Cas9 can be used to generate human VGCC disease mutations in C. elegans, although unexpected phenotypes may result from introducing human mutations in these animals. Nonetheless, this new whole organism model of TS type 1 may provide a foundation for investigating molecular mechanisms involved in this severe genetic disease, screening of additional genetic and therapeutic suppressors as potential treatments for translation, and studying other human channelopathies in C. elegans.
Methods
Request a detailed protocolPlasmid-based CRISPR-Cas9 homologous recombination was performed as previously described (Kim et al. 2014). First, a protospacer adjacent motif (PAM) was identified closest to the desired mutation site (Fig. 1c). Next, custom designed small-guide RNA (sgRNA) oligomers were designed and ordered (IDT) then annealed and ligated into the sgRNA plasmid backbone pRB1017 (Arribere et al. 2014). An 80-nucleotide single stranded oligomer (ssOligo) was custom designed and ordered (IDT) to contain the G-to-R missense mutation, which also co-acted as a silent PAM mutation and XbaI restriction site for efficient PCR screening. The final plasmid injection mix contained 50 ng/µL each of Cas9 vector, pRF4::rol-6(su1006) vector, sgRNA vector, and ssOligo donor. By standard microinjection technique (Mello et al. 1991), a total of 55 wild type animals were injected and 210 F1 animals were cloned then PCR and XbaI screened. This screen yielded three independent heterozygous HR positive lines (~2% efficiency). The G369R allele was maintained by introduction of the DnT1 genetic balancer strain (LGIV), since homozygous TS type 1 animals were unable to produce offspring. This strain (NZ1074) can be requested by contacting D.R.A. at dalbrecht@wpi.edu.
References
Funding
This work is supported by the Burroughs Wellcome Fund, Career Award at the Scientific Interface (D.R.A.), NSF CBET1605679 (D.R.A.) and EF1724026 (D.R.A.), and C.C.M. is a Howard Hughes Medical Institute Investigator.
Reviewed By
Raymond LeeHistory
Received: December 7, 2018Accepted: December 17, 2018
Published: December 18, 2018
Copyright
© 2018 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Lagoy, R; Kim, H; Mello, CC; Albrecht, DR (2018). A C. elegans model for the rare human channelopathy, Timothy syndrome type 1. microPublication Biology. 10.17912/micropub.biology.000081.Download: RIS BibTeX
