
Description
Huntington’s disease (HD) is an autosomal dominant monogenic neurodegenerative disorder caused by a CAG trinucleotide repeat expansion in the gene encoding the protein huntingtin (Htt) (MacDonald et al., 1993). The resultant disease-associated Htt protein harbors a polyglutamine (polyQ) repeat that renders it metastable with respect to folding (Carrell and Lomas, 1997). Htt protein misfolding, characterized by the accumulation of misfolded protein aggregates and neurotoxicity, is first observed in mid- to late-life for most HD patients (Becher et al., 1998). The age-of-onset for HD is inversely proportional to CAG repeat length (Becher et al., 1998). Nonetheless, genetic variation between HD patients is attributed to slight differences in age-of-onset, even when repeat length is the same (Gusella and MacDonald, 2000). Thus, genetic background seems to be an important modifier of Htt protein aggregation and toxicity. We are interested in identifying genes/proteins that enhance or suppress the folding defect of human Htt.
To model Htt toxic-gain-of-function in the genetically tractable Caenorhabditis elegans, we previously characterized transgenic animals expressing a YFP-tagged polyQ-expanded disease-associated fragment of human Htt in C. elegans body wall muscle cells (Lee et al., 2017). More specifically, the first 513 amino acids of the human Htt protein were fused to YFP for visualization. Two different polyQ tract lengths (Q15 and Q128) were utilized, resulting in the proteins Htt513(Q15)::YFP and Htt513(Q128)::YFP, corresponding to the strains EAK102 and EAK103, respectively (Lee et al., 2017). For simplicity, these proteins are referred to herein as Htt513(Q15) or Htt513(Q128). As reported, only Htt513(Q128), not Htt513(Q15), formed protein aggregates in body wall muscle cells (Lee et al., 2017), consistent with only longer polyQ tracts being associated with disease.
Here, we describe the identification and characterization of genetic modifiers of Htt aggregation (mha). To this end, EAK103 animals expressing Htt513(Q128) were grown to the L4 larval stage and exposed to the alkylating agent ethyl methanesulfonate (EMS) at a final concentration of 50mM for 4hrs, according to established protocols (Brenner, 1974). In short, F1 individuals derived from the mutagenized parents were allowed to self-fertilize for one generation, yielding an F2 population, for the purpose of homozygosing recessive alleles and thereby uncovering mutant phenotypes. Screening of the F2 animals for those with increased or decreased aggregation was performed by eye with a fluorescent stereomicroscope.
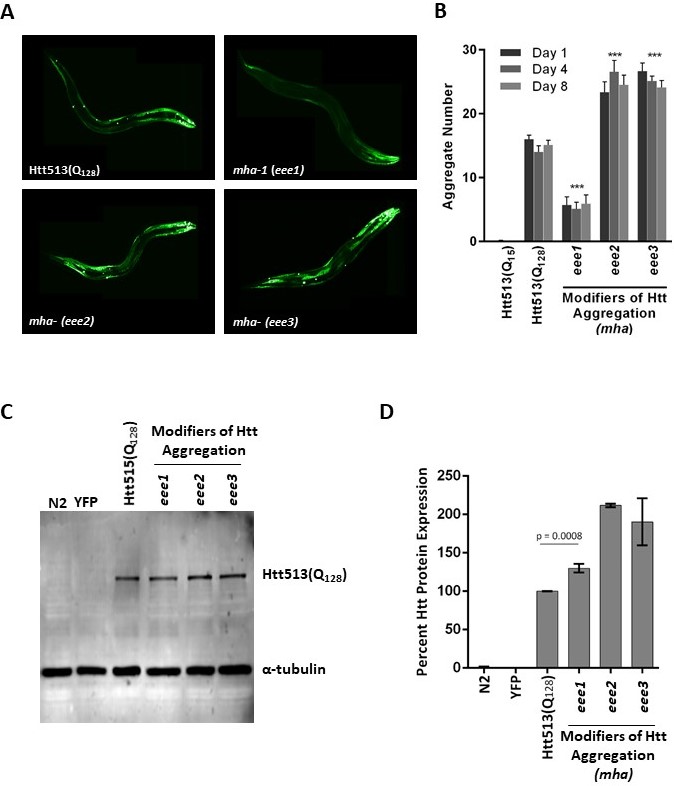
Using this strategy, we obtained three independently-derived mha alleles, mha-1(eee1), mha-(eee2), and mha-(eee3) all displaying either increased or decreased Htt513(Q128) protein aggregation. Independence was assured by generating separate pools of F1 progeny with no more than one mutant individual selected from any given pool for further analysis. However, because allelism tests were not performed, we cannot say that our three alleles necessarily represent three different genes. Therefore, we are assigning only one allele, eee1, the specific gene name mha-1. Qualitatively, mha-1(eee1) displayed decreased Htt513(Q128) aggregation whereas mha-(eee2) and mha-(eee3) displayed increased aggregation (Fig. 1A). To determine the extent of aggregation suppression or enhancement, the number of Htt513(Q128) protein aggregates in each of the three mha mutant strains was quantified at days 1, 4, and 8 of adulthood and compared to that of the Htt513(Q128) parental strain (EAK103) and the Htt513(Q15) negative control strain (EAK102) (Fig. 1B). The aging time-course was to determine whether our new mutants had early or late effects or whether they worked in a synergistic manner with the aging program. We found that while the parental strain accumulated ~15 aggregates in body wall muscle cells on all days examined, mha-1(eee1) accumulated <10 aggregates, whereas mha-(eee2) and mha-(eee3) accumulated >20 aggregates. These numbers of aggregates were statistically different from the parental strain, but not affected by age in any of the mutants examined (Fig. 1B).
Because aggregation is a concentration-dependent phenomenon, we needed to rule out the trivial possibility that changes in aggregation were simply due to higher or lower transgene expression levels. To address this, we performed western blot analysis with an antibody raised against expanded polyQ or against α-tubulin as a loading control (Fig. 1C). Briefly, total protein from animals grown to day 1 of adulthood was extracted and loaded on a 10% SDS-polyacrylamide gel, transferred to a PVDF membrane, and incubated in the presence of the indicated antibodies. Visualization was with a Li-Cor Odyssey imaging system (Lincoln, NE). Quantification of protein levels from three independent experiments was performed with Image J. The analysis revealed that mha-1(eee1) accumulated more, not less, Htt513(Q128) protein than the parental control (Fig. 1D). This means that the underlying genetic lesion in mha-1(eee1) decreases aggregation without decreasing protein levels. In contrast, mha-(eee2) and mha-(eee3), in which Htt513(Q128) aggregated more than the control, also accumulated more total protein. Thus, the observed increase in aggregation could be due to a higher concentration of available Htt513(Q128) protein. Alternatively, these aggregates themselves may be highly stable, such that increased aggregation may equate to less protein turnover, a longer half-life, and, consequently, higher steady-state protein levels.
Together, our data describe a screening strategy for the successful identification of genetic modifiers of Htt513(Q128) protein aggregation. Prior to this study, we were uncertain whether the long polyQ tract length and early aggregation of Htt513(Q128) in body wall muscle cells would render it impossible to suppress aggregation. However, mha-1(eee1) only forms a few aggregates in the muscles surrounding the head, but is otherwise completely diffuse. Thus, the mutants characterized herein are not only interesting in their own right as modifiers of protein aggregation, but they serve as a proof of principle, opening up the possibility for larger-scale studies in the future.
Reagents
C. elegans were maintained on Nematode Growth Media (NGM) that was seeded with E. coli strain OP50 as a food source according to established protocols (Brenner 1974).
The mha mutant strains described herein are available by request. They are:
EAK104 mha-1(eee1) eeeIs2[Punc-54::Htt513(Q128)::YFP::unc-54 3’UTR]
EAK105 mha-(eee2) eeeIs2[Punc-54::Htt513(Q128)::YFP::unc-54 3’UTR]
EAK106 mha-(eee3) eeeIs2[Punc-54::Htt513(Q128)::YFP::unc-54 3’UTR]
The following previously published strains used in this study are available from the C. elegans Genetic Center (CGC):
N2 Wild type, Bristol, (Brenner 1974)
AM134 rmIs126[Punc-54::Q0::YFP] X, (Morley, Brignull et al. 2002)
EAK102 eeeIs1[Punc-54::Htt513(Q15)::YFP::unc-54 3’UTR], (Lee, Ung et al. 2017)
EAK103 eeeIs2[Punc-54::Htt513(Q128)::YFP::unc-54 3’UTR], (Lee, Ung et al. 2017)
For SDS-PAGE and western blot analysis, total protein from 10 nematodes was extracted directly into Laemmli sample buffer and loaded on a 10% SDS-PAGE gel. After electrophoresis, protein was transferred to an Immun-Blot Low Fluorescence PVDF membrane (Bio-Rad, Irvine, CA). The primary antibodies were the anti-expanded polyglutamines antibody 3B5H10 and the anti-alpha-tubulin antibody B-5-1-2, both from Millipore Sigma (Carlsbad, CA). The secondary antibody was the IRDye® 800CW Goat anti-Mouse IgG Secondary Antibody from Li-Cor (Lincoln, NE).
Acknowledgments
We thank members of the Kikis lab, especially Emily Green, for technical support. Additionally, this research project was supported in part by the Emory University Integrated Cellular Imaging Microscopy Core.
References
Funding
EAK was supported by a Faculty Fellowship from the Appalachian College Association and by a Faculty Development Grant from the University of the South.
Reviewed By
Cindy VoisineHistory
Received: December 6, 2019Accepted: December 20, 2019
Published: January 2, 2020
Copyright
© 2020 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Ung, HM; Hall, RH; Kikis, EA (2020). Chemical mutagenesis of Caenorhabditis elegans uncovers genetic modifiers of huntingtin protein aggregation. microPublication Biology. 10.17912/micropub.biology.000202.Download: RIS BibTeX
