University of Rochester, Department of Medicine, Nephrology Division, Rochester, NY

Description
Alzheimer’s disease (AD) is the most common progressive neurodegenerative disorder (Selkoe et al., 2001). One of the key pathological hallmarks of AD is neurofibrillary tangles (NFTs), which are primarily composed of abnormally modified tau (Avila et al., 2004). Tau isolated from AD brain exhibits a number of posttranslational modifications (PTMs); including increases in phosphorylation and acetylation at specific epitopes that likely impair its function (Neddens et al., 2018). Phosphorylation of tau at threonine 231 (T231) causes significant changes in tau structure, thus impairing microtubule binding (Mi et al., 2006; Quintanilla et al., 2014). In addition, increased expression of tau acetylated at Lysine 274 (K274) and Lysine 281 (K281) appears to result in mislocalization of tau, destabilization of the cytoskeleton in the axon initial segment, and synaptic dysfunction (Tracy et al., 2016). Even though it is widely accepted that tau with aberrant PTMs facilitate neurodegeneration, the precise cellular mechanisms remain unknown. Mounting evidence suggests selective pathological tau species compromise mitochondrial biology (Reddy et al., 2011; Cummins et al., 2019). Understanding the molecular mechanisms through which this occurs will help to delineate the role tau plays in AD. Mitochondrial quality control mechanisms play a key role in restoring cellular homeostasis following stress. In addition, these mechanisms promote mitochondrial recycling through a form of selective autophagy termed mitophagy, are an attractive target to consider in the context of AD (Kerr et al., 2017).
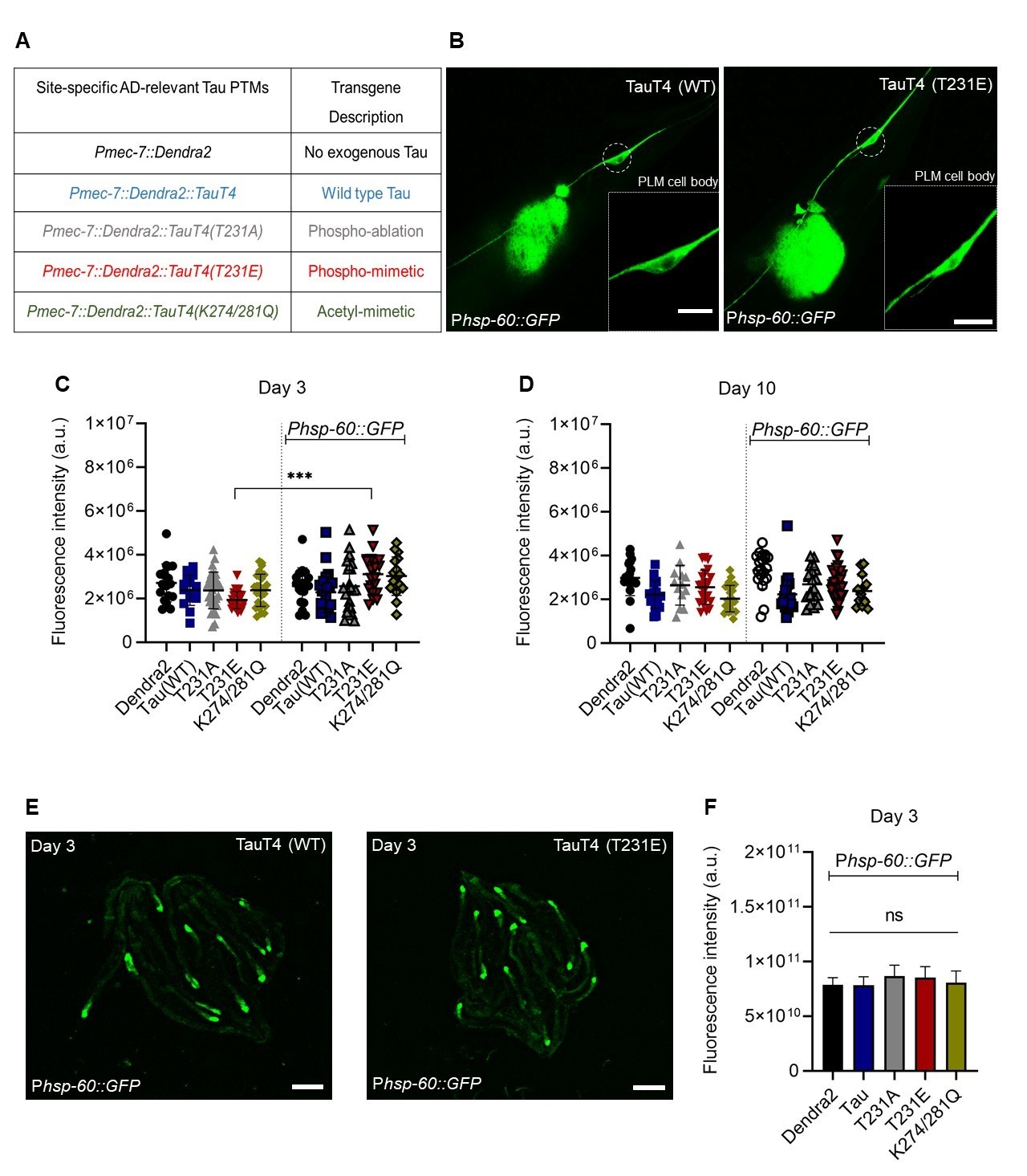
To interrogate the effects of pathologic PTMs in Caenorhabditis elegans model system, CRISPR-Cas9 gene editing (Paix et al., 2015) was used to introduce a disease-associated phosphorylation mimicking (T→E) or a non-phosphorylatable (T→A) mutation at the T231 position of the wild-type TauT4 isoform, or alternatively acetylation mimicking (K→Q) mutations at the K274 and K281 positions, as listed in Fig. 1A. Overall, our results clearly demonstrated that the induction of mitophagy occurring in response to the mitochondrial toxin paraquat was entirely suppressed by expression of the T231E and K274/281 mutants, but not of wild type TauT4 itself (Guha et al., 2020).
One intriguing possibility is that these two TauT4 PTM-mimetic mutants might induce a mild adaptive stress response during development that dampens subsequent responsiveness following overt stress. In fact, it has been well documented that many signaling pathways that sense stress have feedback loops to suppress their sustained activation (Hotamisligil et al., 2016), supporting at least plausibility. The mitochondrial unfolded protein response (UPRmt) is one such pathway. The UPRmt is a surveillance pathway that was first identified in mammals, but has been best characterized genetically in the nematode C. elegans (Haynes et al., 2007; Melber et al., 2018). Induction of UPRmt initiates a mitochondria-nuclear signaling axis that protects against stresses caused by respiratory chain deficits, excessive reactive oxygen species, unfolded proteins, and pathologic bacteria (Rolland et al., 2019; Peña et al., 2016). In C. elegans, HSP-60 is a matrix-localized mitochondrial molecular chaperone whose expression has been widely used as a surrogate for activation of the UPRmt (Bennett et al., 2014; Benedetti et al., 2006). The expression of an integrated transgene where GFP is driven by the hsp-60 promoter is restricted under basal condition to the posterior cells of the intestine, but following induction of the UPRmt is expressed more widely throughout the body (Gitschlag et al., 2016). Here, we asked whether the expression of Phsp-60::GFP in touch neurons is influenced by different PTMs of tau.
The observation that measurable green fluorescence could be observed in the cell body of PLM touch neurons even in the absence of the Phsp-60::GFP transgene (Fig. 1B and 1C), is consistent with the fluorescent spectra of the photo-convertible Dendra2 tag, which is present in all of the transgenic strains, albeit at single copy. However, in strains containing the Phsp-60::GFP transgene, only the phospho-mimetic T231E mutant exhibited a significant increase in PLM cell body fluorescence (Fig. 1C). The magnitude of the response is small, but this result is consistent with T231E causing mild mitochondrial stress and cell-autonomous activation of the UPRmt. Interestingly, UPRmt activation in the K274/281Q acetyl-mimetic mutant, which like T231E caused neurodegeneration and an inability to trigger mitophagy in response to paraquat treatment (Guha et al., 2020), failed to reach significance (p = 0.1), but also had increased scatter in the baseline signal (Fig. 1C).
One advantage of the worm model with its short three weeks lifespan is the ability to relate longitudinal effects over time to aging. In this context we note that the increased Phsp-60::GFP expression in T231E was limited to day 3 of adulthood, and that the difference between fluorescence in the absence versus the presence of the Phsp-60::GFP transgene was not significant at day 10 in any of the strains (Fig. 1D). While this may have been due to the limited magnitude of the response and a higher baseline fluorescence in older animals (Fig. 1D), it is also possible that it reflects the fact that the UPRmt is thought to be restricted to young animals (Wu et al., 2018).
Another distinct advantage of the worm model is the ability to discern between cell autonomous and cell non-autonomous activation of stress signaling pathways, a mode of activation relevant to both the UPRmt (Durieux et al., 2011) and the more widely studied UPRer in worms (Frakes et al., 2017). However we were unable to detect any change in intestinal Phsp-60::GFP expression across the entire repertoire of tau mutants (Fig. 1E, F), suggesting that the increased expression we observed in the T231E mutant was limited to a cell autonomous effect.
Our results clearly demonstrate that wild type tau expressed at single copy level does not activate the UPRmt in worm touch neurons, but that a mutation mimicking a pathological PTM that has been associated with AD causes subtle UPRmt activation in young adult worms. We hypothesize that this chronic low-level activation could suppress subsequent responses to mitochondrial stress.
Methods
Request a detailed protocolC. elegans strains growth and maintenance
Nematodes were maintained at 200C on Nematode Growth Media (NGM) plates made with Bacto Agar (BD Biosciences). The plates were seeded with live E. coli OP50-1 bacterial strain (cultured overnight at 37oC at 220 rpm) and allowed to grow overnight. For experimental assays, after synchronization by standard procedure with sodium hypochlorite, 4th larval stage (L4) hermaphrodites (characterized by the appearance of a “Christmas tree vulva”) were selected and moved to test plates. The day after moving was considered adult day 1, and animals were assayed on day 3 and day 10. Animals were transferred daily to avoid mixed population until they stop laying eggs.
Fluorescent imaging assay
Animals were mounted on 2% agarose pads on glass slides and immobilized with 1 mM tetramisole hydrochloride before imaging. Imaging was performed using a Nikon Eclipse inverted microscope coupled to a six channel LED light source (Intelligent Imaging Innovation, Denver, CO), an ORCA-Flash4.0 V2 Digital CMOS camera (Hamamatsu Photonics, Bridgewater Township, NJ) and Slidebook6 software (Intelligent Imaging Innovation, Denver, CO). All images were acquired under the same exposure conditions and each experiment was imaged in one session. The PLM cell body was identified by their position toward the posterior of the animal, near the tail and was focused with a 100x oil immersion lens under visible light using DIC contrast. 600-nm+ emissions were captured first following excitation at 440-nm, keeping light intensity and exposure times constant between images. Images were quantified using ImageJ software by selecting the ROI, measuring the mean intensity for green channels and subtracting the background intensity. N.B. – We only quantified PLM cell body fluorescence, not the ALM fluorescence because it might interfere with the intestinal gut fluorescence, giving us a faulty reading.
Statistical Analysis
All statistical analyses were conducted using Prism 8.0 (GraphPad Software), with alpha-error level of p < 0.05 considered to be significant. Data were averaged and represented as mean ± standard deviation (mean ± SD). In general, group differences were analyzed with either one-way or two-way ANOVA depending upon the variables. The sample sizes were based on those found previously in the laboratory to provide appropriate power for discerning phenotypic differences among genotypes.
Reagents
SJ4058: zcIs9 [hsp-60::GFP + lin-15(+)]. This is a stable transgenic line with low basal GFP expression, mainly in the tail, observed from L1 to adult animals. Transgenic strains include the following: KWN169, rnySi26 [Pmec-7::Dendra2; unc-119+] II; KWN167, rnySi24 [Pmec-7::Dendra2::Tau-T4; unc-119+] II. KWN788 rnySi51 [Tau-T4 (T231A) *rnySi24] II, KWN789 rnySi52 [Tau-T4 (T231E) *rnySi24] II, KWN790 rnySi53 [Tau-T4 (K274Q; K281Q) *rnySi24] II. For crossing tau MosSCI strains into hsp-60 reporter strain, Dendra2 fluorescent was used to guide selection of homozygous mutants, and PCR genotyping was used to confirm homozygosity with primers specific to the ttTi5605 loci, including:
MosSCI ttTi5605-F, 5’GTTTTTGATTGCGTGCGTTA3’
MosSCI ttTi5605-R, 5’ACATGCTTCGTGCAAAACAG3’
MosSCI ttTi5605 insert-F, 5’CATCCCGGTTTCTGTCAAAT3’
Acknowledgments
Acknowledgements Technical assistance provided by Joseph Cartella and Alan Alberto was greatly appreciated. We thank the members of Dr. Johnson’s lab, the Mitochondrial Research and Interest Group at the University of Rochester Medical Center and the members of the Western New York worm meeting for their valuable suggestions and helpful discussions.
References
Funding
R21 AG060627. R01AG067617.
Reviewed By
AnonymousHistory
Received: August 23, 2020Revision received: September 4, 2020
Accepted: September 6, 2020
Published: September 9, 2020
Copyright
© 2020 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Guha, S; Fischer, S; Cheng, A; Johnson, GV; Nehrke, K (2020). A T231E Mutant that Mimics Pathologic Phosphorylation of Tau in Alzheimer’s disease Causes Activation of the Mitochondrial Unfolded Protein Response in C. elegans touch neurons. microPublication Biology. 10.17912/micropub.biology.000306.Download: RIS BibTeX
