Robert J. and Nancy D. Carney Institute for Brain Science, Providence, RI 02912
Neuroscience Graduate Program, Brown University, Providence, RI 02912

Description
Amyotrophic lateral sclerosis (ALS) is a fatal degenerative motor neuron disease. While the mechanisms underlying motor neuron death in ALS are not well understood, mutations in over 25 genes can cause this disease (Marangi and Traynor 2015). It remains unclear which, if any, of these genes act in the same disease-associated pathway(s), or if they act in the same pathway(s) as genes associated with the related disorder, frontotemporal dementia (FTD) (Ling et al. 2013). The first ALS-causing gene to be identified was superoxide dismutase 1 (SOD1), a regulator of cytoplasmic redox homeostasis (Rosen et al. 1993). We can begin to construct a pathway for neurodegeneration through SOD1 by identifying genes whose loss of function (LOF) modifies the level of degeneration in a C. elegans SOD1 ALS model. This will contribute to our understanding of whether ALS/FTD genes act in a single or multiple pathways to cause disease.
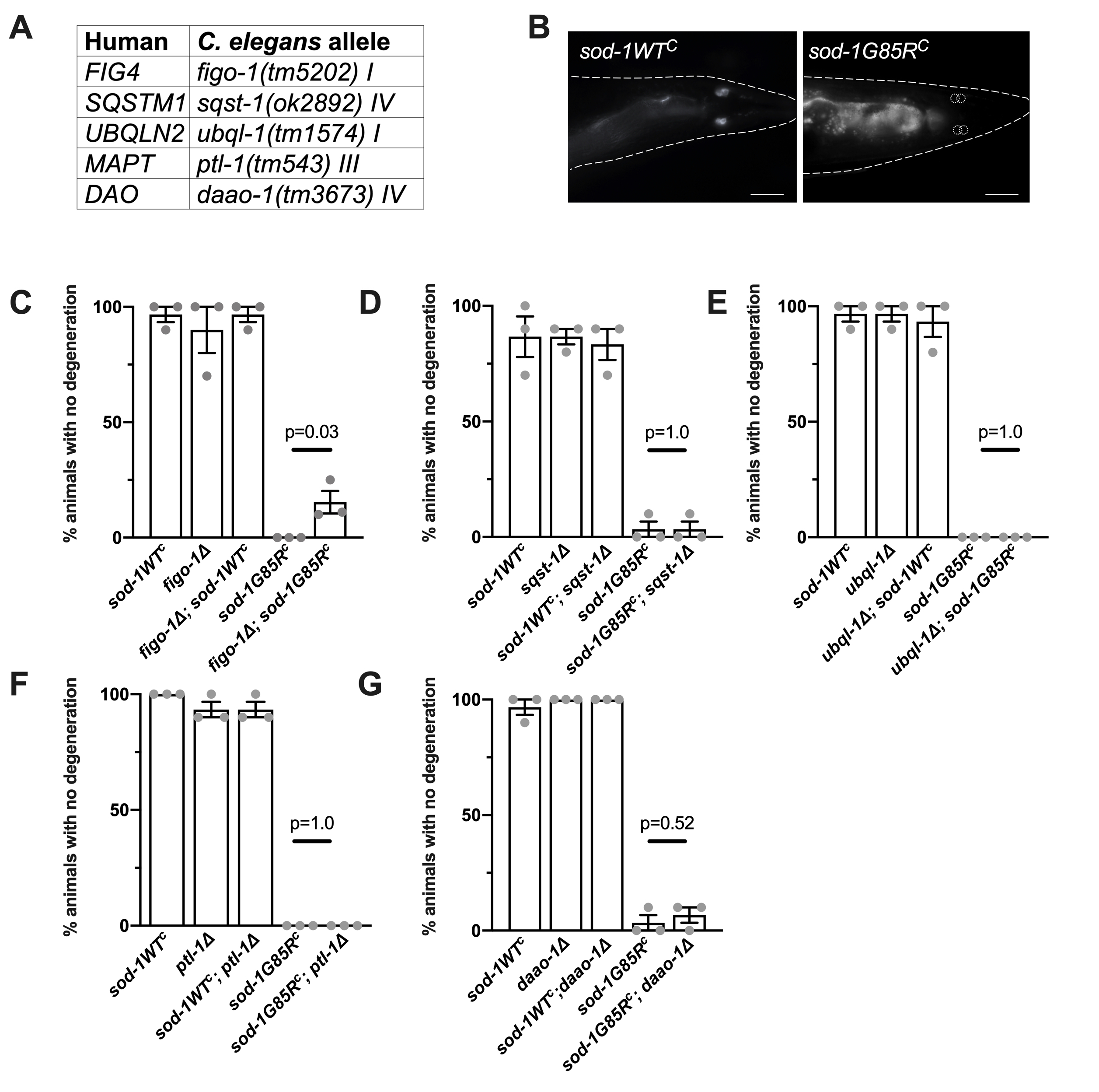
To undertake this analysis, we introduced LOF alleles for C. elegans orthologs of ALS or FTD genes into a C. elegans knock-in model of SOD1 ALS (Baskoylu et al. 2018). Previously, we used CRISPR/Cas9 to introduce a point mutation corresponding to the ALS-causing SOD1 G85R variant (Rosen et al. 1993) into a conserved residue in the endogenous C. elegans sod-1 gene (Baskoylu et al. 2018). sod-1(rt448) contains the G85R mutation and sod-1(rt449) is the corresponding control strain containing silent edits; these are referred to herein using the corresponding human patient allele nomenclature, sod-1G85RC and sod-1WTC (Baskoylu et al. 2018). sod-1G85RC animals exhibit glutamatergic neuron degeneration following exposure to mild oxidative stress. To begin exploring genetic interactions in this model, we selected several genes with existing deletion alleles (Figure 1A); deletion alleles were crossed into the sod-1G85RC background to investigate their effect on sod-1G85R glutamatergic neuron degeneration. ALS/FTD-causing gene orthologs tested include: figo-1, ortholog of factor-induced gene 4 (FIG4), a regulator of PI(3,5)P2, a phosphoinositide phosphatase involved in endosomal signaling and trafficking (Chow et al. 2009); sqst-1, an ortholog of human sequestosome 1 (SQSTM1), which encodes the SQSTM1/p62 cargo protein involved in selective autophagy (Fecto et al. 2011); ubql-1, ortholog of ubiquilin-2 (UBQLN2), a carrier in the ubiquitin/proteasome system (Deng et al. 2011); ptl-1, ortholog of microtubule-associated protein tau (MAPT), a player in microtubule assembly and dynamics also implicated in Alzheimer’s disease and various dementias (Fang et al. 2013, Rademakers et al. 2004); and daao-1, ortholog of D-amino acid oxidase (DAO), among whose substrates is D-serine, an endogenous neurotransmitter and co-agonist of NMDA receptors (Mitchell et al. 2010).
Loss of glutamatergic neurons was quantified by dye-filling four glutamatergic neurons (PHAR/L and PHBR/L) in the tail with exposed sensory endings (Figure 1B). After mild oxidative stress with paraquat (22 hours, 2.5mM), the majority of these neurons in sod-1G85RC animals are unable to take up dye and are presumed to have degenerated (Baskoylu et al. 2018). figo-1(tm5202); sod-1G85RC animals exhibited increased dye-filling compared to sod-1G85RC (Figure 1C), indicating loss of figo-1 partially suppresses glutamatergic neuron degeneration in sod-1G85RC animals. sod-1G85RC; sqst-1(ok2892), ubql-1(tm1574); sod-1G85RC, sod-1G85RC; ptl-1(tm543), and sod-1G85RC; daao-1(tm3673) animals exhibited no significant difference in dye-filling compared to sod-1G85RC (Figure 1D-G). LOF alleles did not show stress-induced neuron degeneration in combination with sod-1WTC (Figure 1C-G).
Based on these results, figo-1 and sod-1 may lie in the same genetic pathway. sod-1 LOF is predominantly responsible for glutamatergic neuron degeneration in sod-1G85RCanimals (Baskoylu et al. 2018). As figo-1 LOF suppressed glutamatergic neuron degeneration in the sod-1G85RCmodel, figo-1 likely lies downstream of sod-1. The epistatic relationship observed here will need to be confirmed with other deletion alleles of figo-1 before potential mechanisms of FIG4 and SOD1 interaction are investigated. figo-1 may make neurons more robust or specifically resistant to sod-1-related glutamatergic neuron loss. Additionally, SOD1 ALS-related alterations in endosomal activity have been reported (van Dis et al. 2014; Xie et al. 2015) and may be influenced by the indirect role of FIG4 in endosomal signalling and trafficking. Further examination of epistatic relationships between SOD1 and other ALS/FTD-causing genes can yield a complete pathway of SOD1 action, as well as elucidate whether ALS/FTD is a single disease consisting of one or more pathways or perhaps even multiple diseases with shared characteristics.
Methods
Request a detailed protocolC. elegans strains
All strains were maintained using standard methods (Brenner 1974) and constructed using the C. elegans Bristol variety N2 as a wild-type parent strain. All strains were backcrossed to N2 at least 4 times before use.
List of C. elegans strains (Strain name: genotype)
• HA2986: sod-1(rt448[WTC]) II; pha-1(+) III (abbreviated sod-1WTC)
• HA3299: sod-1(rt449[G85RC]) II; pha-1(+) III (abbreviated sod-1G85RC)
• HA3261: figo-1(tm5202) I
• HA3266: sqst-1(ok2892) IV
• HA3352: ubql-1(tm1574) I
• HA3402: ptl-1(tm543) III
• HA3361: daao-1(tm3673) IV
• HA3788: figo-1(tm5202) I; sod-1WTC II
• HA3789: figo-1(tm5202) I; sod-1G85RC II
• HA3509: sod-1WTC II; sqst-1(ok2892) IV
• HA3510: sod-1G85RC II; sqst-1(ok2892) IV
• HA3516: ubql-1(tm1574) I; sod-1WTC II
• HA3517: ubql-1(tm1574) I; sod-1G85RC II
• HA3996: sod-1WTC II; ptl-1(tm543) III
• HA3997: sod-1G85RC II; ptl-1(tm543) III
• HA3795: sod-1WTC II; daao-1(tm3673) IV
• HA3796: sod-1G85RC II; daao-1(tm3673) IV
• FX19472: tmIn10 [mIs14 spy-10(e128)] II, used in strain construction
Strains are available upon request to anne_hart@brown.edu.
Dye-filling assay
To quantify loss of glutamatergic sensory neurons, we followed a procedure outlined in Perkins et al. 1986. Animals raised to larval stage 4 were transferred to plates containing 2.5 mM paraquat (Acros Organic, #1910-42) for 22 hours at 25C with minimal light exposure. After the incubation period, 2 mg/mL DiI (Fisher DiIC18(5) D307) was added. After 1.5 hours, animals were transferred back to regular NGM plates, then immobilized with 30 mg/mL 2-3-butanedione monoxime (BDM, Sigma) in M9 buffer and mounted on 2% (vol/vol) agar pads. Fluorescent neuronal cell bodies in the tail were counted under 63x objective on a Zeiss Axioplan2. Animals with three or four phasmid neurons that fill with dye were scored as having no degeneration (Figure 1B).
Acknowledgments
Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), and the National BioResource Project (NBRP, Japan). We thank members of the Hart lab and Dr. Mark Johnson for useful discussions.
References
Funding
This work was supported by a Karen T. Romer Undergraduate Teaching and Research Award (JFO), by NIMH (T32MH20068) and NINDS (T32NS62443) training grants to the Neuroscience Department Program at Brown University (KSY), and by an ALS Association (wa111) and Brown Institute for Brain Science Innovation Award (ACH).
Reviewed By
Alex ParkerHistory
Received: November 13, 2020Revision received: December 22, 2020
Accepted: January 9, 2021
Published: January 15, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Osborne, JF; Yanagi, KS; Hart, AC (2021). Genetic interactions in a C. elegans sod-1 ALS model: glutamatergic neuron degeneration. microPublication Biology. 10.17912/micropub.biology.000338.Download: RIS BibTeX
