Department of Biology, Brandeis University, Waltham, MA
RIKEN Hakubi Research Team, RIKEN Cluster for Pioneering Research, RIKEN Center for Brain Science, Wako, Japan

Abstract
Degenerate networks can drive similar circuit outputs. Via acute manipulation of individual neurons, we previously identified circuit components that are necessary and sufficient to drive starvation-dependent plasticity in C. elegans thermotaxis behavior. Here we find that when these components are instead silenced chronically, degenerate mechanisms compensate to drive this behavior. Our results indicate that degeneracy in neuronal network function can be revealed under specific experimental conditions.
Description
Neuronal networks can achieve similar outputs via distinct underlying circuit mechanisms (Beverly et al., 2011; Marder et al., 2015; Saideman et al., 2007; Trojanowski et al., 2014; Wang et al., 2019). This degeneracy allows networks to maintain robustness without compromising functional flexibility (Cropper et al., 2016; Edelman and Gally, 2001). Since the contribution of degenerate neuronal pathways is likely to be revealed under defined genetic or environmental conditions, it is challenging to identify and describe the contributions of such pathways to neuronal circuit function.
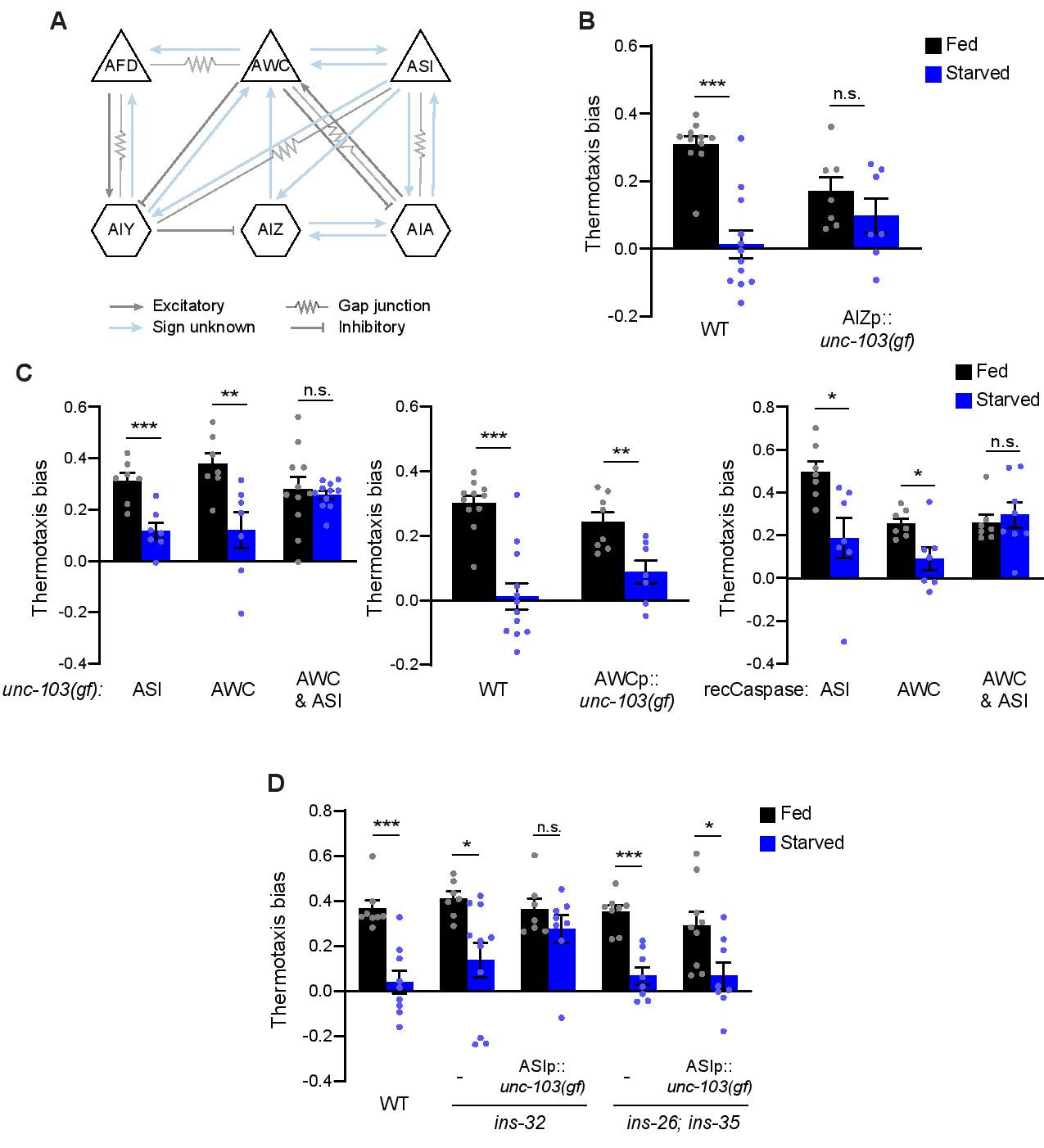
C. elegans exhibits experience-dependent thermotaxis behaviors (Hedgecock and Russell, 1975). When animals are placed on a thermal gradient at temperatures warmer than the temperature at which they were cultivated with bacterial food (cultivation temperature – Tc), they move towards cooler temperatures (negative thermotaxis – NT). Genetic ablation experiments established that in well-fed animals, the AFD, AWC and ASI sensory neuron pairs act degenerately to drive NT under specific assay conditions (Beverly et al., 2011; Ikeda et al., 2020; Kuhara et al., 2008; Mori and Ohshima, 1995). Starvation for 2-3 hrs prior to the assay disrupts NT, resulting in atactic behavior (Chi et al., 2007; Hedgecock and Russell, 1975; Kodama et al., 2006) (referred to here as ‘starvation-dependent plasticity’). Via acute chemogenetic and optogenetic manipulation of neuron activity during the behavioral assay (henceforth referred to as ‘acute silencing’), we recently showed that increased and decreased temperature responses in AWC and their postsynaptic AIA interneuron partners (Figure 1A), respectively, are necessary and sufficient for starvation-dependent plasticity in NT (Takeishi et al., 2020). In the course of these experiments, we noted that while temperature responses in the AIZ interneurons (Figure 1A) were also modulated by starvation in a manner similar to responses in AIA, acute chemogenetic manipulation of AIZ activity had no effect on NT regardless of feeding conditions (Takeishi et al., 2020). We speculated that AIZ may be a component of a degenerate thermotaxis behavioral circuit that contributes to starvation-dependent plasticity in NT under specific conditions.
We tested whether silencing AIZ throughout postembryonic development (henceforth referred to as ‘chronic silencing’) may reveal a role for this interneuron type in modulating thermotaxis. While histamine-HisCl1-mediated acute silencing of AIZ had no effect on NT (Takeishi et al., 2020), chronic silencing via expression of an activated UNC-103 potassium channel [unc-103(gf)] (Reiner et al., 2006) in this neuron significantly decreased NT regardless of feeding conditions (Figure 1B), possibly due to disruption of AIZ-mediated regulation of reversals and turns. In contrast, both acute and chronic silencing of AIA abolished NT similarly in fed and starved animals (Takeishi et al., 2020). These results indicate that while AIA is able to drive NT when AIZ is acutely silenced, AIA is unable to compensate when AIZ is silenced chronically.
We next examined whether the contributions of sensory neurons upstream of AIA and AIZ (Figure 1A) to NT are also distinct based on the duration and/or timing of neuronal silencing. Acute chemogenetic or optogenetic silencing of AWC alone during thermotaxis abolished starvation-dependent plasticity in NT without affecting NT in fed animals (Takeishi et al., 2020). Unexpectedly, we found that starved animals in which AWC has been chronically silenced via expression of unc-103(gf) or genetic ablation (Beverly et al., 2011; Chelur and Chalfie, 2007), continued to exhibit starvation-dependent plasticity in NT similar to the behavioral phenotype of wild-type animals (Figure 1C), but in stark contrast to the behavior of animals in which AWC is acutely silenced (Takeishi et al., 2020). To confirm that the observed behavioral differences were not simply due to differences in the promoters used for chronic and acute silencing (see Methods), we also expressed unc-103(gf) under odr-1 regulatory sequences; odr-1 promoter sequences were used previously to drive HisCl1 and GtACR2 expression in AWC (Takeishi et al., 2020). Animals expressing odr-1p::unc-103(gf) again exhibited starvation-dependent plasticity in NT (Figure 1C). The NT behavioral phenotypes of animals in which ASI was acutely or chronically silenced resembled those of wild-type animals (Figure 1C) (Takeishi et al., 2020). We found that chronic silencing of both AWC and ASI together was necessary to fully abolish starvation-dependent plasticity in NT (Figure 1C), phenocopying the effects of silencing AWC alone acutely (Takeishi et al., 2020). These results indicate that ASI can contribute to starvation-dependent plasticity in NT, but that a role for this neuron is only revealed when AWC is chronically but not acutely silenced.
We previously noted that in contrast to the lack of an effect of chronic silencing of AWC alone on starvation-dependent plasticity in NT, chronically blocking glutamatergic transmission from AWC was sufficient to abolish this plasticity, similar to the effects observed upon acutely silencing AWC alone (Takeishi et al., 2020). A simple model to account for these seemingly contradictory observations is that AWC also releases a peptidergic signal upon starvation. Absence of this signal due to chronic silencing of AWC, but not upon acute AWC silencing or upon loss of glutamatergic signaling from AWC, may trigger ASI-dependent compensatory mechanisms. This model predicts that a loss-of-function mutation in such a peptide-encoding gene would phenocopy the effects of AWC chronic silencing on starvation-dependent plasticity in NT.
We reported a subset of insulin-like peptide (ILP) genes expressed in AWC which plays a role in transmitting information about food availability to influence the dauer developmental decision (Neal et al., 2015). We tested whether one or more of these ILP genes may play a role in starvation-dependent plasticity in NT. The NT behavioral phenotypes of ins-32 mutants alone or ins-26; ins-35 double mutants were similar to those of wild-type animals in both fed and starved conditions (Figure 1D). However, while the NT behavioral phenotype of ins-26; ins-35 mutants was not further altered upon ASI chronic silencing, chronic silencing of ASI in ins-32 mutants abolished starvation-dependent plasticity in NT (Figure 1D). Thus, in this context, loss-of-function mutations in ins-32 appear to phenocopy the effects of chronic AWC silencing.
We propose the following model to account for observations reported here and in our previous work (Takeishi et al., 2020). Long-term chronic silencing of AWC may promote compensatory mechanisms via ASI to mediate starvation-dependent plasticity in NT. This compensation may be triggered by the prolonged absence of ins-32 ILP signaling from AWC. Although the nature of the proposed ASI-dependent compensatory pathway is currently unclear, altered peptidergic signaling from ASI may play a role (Chen et al., 2013). Acute AWC silencing during the behavioral assay alone may not be sufficient to induce this ASI-dependent compensatory mechanism. Alternatively, the relevant compensatory mechanism may be absent in adult animals. Developmental compensation in C. elegans neuronal networks has previously been reported and has been suggested to potentially be mediated via rewiring of synaptic connections or altered functions of other circuit components (Beverly et al., 2011; Trojanowski et al., 2014; White et al., 2007). Rewiring may account for the differential requirements of AIA and AIZ in driving NT upon chronic and acute silencing. However, given the extensive peptidergic and hormonal signaling network in C. elegans, we instead favor the hypothesis that this compensation and degeneracy instead arises from reconfiguration of the ‘wireless’ connectome (Bargmann and Marder, 2013; Bentley et al., 2016). Our results further reinforce that conclusions drawn about neuron circuit components driving specific behaviors should be interpreted with caution in the context of the specific manipulations performed.
Methods
Request a detailed protocolC. elegans growth
C. elegans was maintained at 20°C with E. coli OP50 as the food source. To generate transgenic animals, 5-30 ng/μl of experimental plasmids were injected with co-injection markers (unc-122p::gfp, unc-122p::dsRed, elt-7p::gfp, or elt-7p::mCherry). Mutations were confirmed by sequencing. Strains used in this work are listed in Table 1.
Molecular biology
Promoters used in this work were (upstream of ATG): odr-1 (AWC: 1.0 kb), ceh-36delp (AWC: 627 bp), srg-47 (ASI: 650 bp), odr-2b(3a) (AIZ and others: 448 bp), ser-2(2) (AIZ and others: 4.7 kb). Promoters and cDNA sequences were cloned into the pPD95.77 expression vector. All plasmids were confirmed by sequencing. Plasmids used in this work are listed in Table 2.
Thermotaxis behavioral assay
Negative thermotaxis behavioral assays were performed as previously described (Takeishi et al., 2020). Starved worms were obtained by transferring animals onto unseeded NGM plates for 3 hrs prior to the start of the assay. Assays were performed on a thermal gradient ranging from 23°C-28°C at 0.5°C/cm.
Reagents
| Table 1. Strains used in this work. | ||
| Strain | Genotype | Source |
| Wild type | N2 (Bristol) | CGC |
| PY12216 | oyEx676 [ser-2(2)p::FRT::STOP::FRT::unc-103(gf)::SL2::gfp + odr-2b(3a)p::nFLP + elt-7p::gfp] | This work |
| PY12220 | oyIs92 [ceh-36delp::unc-103(gf)::SL2::mCherry + unc-122p::gfp] | This work |
| PY7502 | oyIs85 [ceh-36delp::TU813 + ceh-36delp::TU814 + unc-122p::dsRed + srtx-1p::gfp] | (Beverly et al., 2011) |
| PY12221 | oyIs93 [srg-47p::unc-103(gf)::SL2::mCherry + elt-7p::mCherry] | This work |
| PY7505 | oyIs84 [gpa-4p::TU813 + gcy-27p::TU814 + gcy-27p::gfp + unc-122p::dsRed] | (Beverly et al., 2011) |
| PY7507 | oyIs84 [gpa-4p::TU813 + gcy-27p::TU814 + gcy-27p::gfp + unc-122p::dsRed]; oyIs85[ceh-36delp::TU813 + ceh-36delp::TU814 + unc-122p::dsRed + srtx-1p::gfp] | (Beverly et al., 2011) |
| PY12223 | oyIs93 [srg-47p::unc-103(gf)::SL2::mCherry + elt-7p::mCherry]; oyIs92 [ceh-36delp::unc-103(gf)::SL2::mCherry + unc-122p::gfp] | This work |
| PY12217 | oyEx677 [odr-1p::unc-103(gf)::SL2::mCherry + unc-122p::gfp] | This work |
| PY10712 | ins-26(tm1983); ins-35(ok3297) | (Neal et al., 2015) |
| PY12219 | ins-26(tm1983); ins-35(ok3297); oyIs93 [srg-47p::unc-103(gf)::SL2::mCherry + elt-7p::mCherry] | This work |
| PY10808 | ins-32 (tm6109) | (Neal et al., 2015) |
| PY12218 | ins-32 (tm6109); oyIs93[srg-47p::unc-103(gf)::SL2::mCherry + elt-7p::mCherry] | This work |
| Table 2. List of plasmids used in this work. | ||
| Plasmid | Description | Source |
| PSAB1255 | ser-2(2)p::FRT::STOP::FRT::unc-103(gf)::SL2::gfp | This work |
| PSAB1253 | ceh-36delp::unc-103(gf)::SL2::mCherry | This work |
| PSAB1254 | srg-47p::unc-103(gf)::SL2::mCherry | This work |
| PSAB1256 | odr-1p::unc-103(gf)::SL2::gfp | This work |
| pWY046 | odr-2b(3a)p::nFLP | (Takeishi et al., 2020) |
Acknowledgments
We thank Wenxing Yang for sharing reagents, the Caenorhabditis Genetics Center for strains, and Nathan Harris and the Sengupta lab for advice and comments on the manuscript.
References
Funding
This work was partly supported by the Japan Society for the Promotion of Science (H28-1058 – A.T.) and the NIH (R35 GM122463 – P.S.).
Reviewed By
AnonymousHistory
Received: January 5, 2021Revision received: January 8, 2021
Accepted: January 11, 2021
Published: January 15, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Yeon, J; Takeishi, A; Sengupta, P (2021). Chronic vs acute manipulations reveal degeneracy in a thermosensory neuron network. microPublication Biology. 10.17912/micropub.biology.000355.Download: RIS BibTeX
