Department of Neurobiology, Northwestern University

Abstract
Accumulating evidence demonstrates that mutations in ALDH1A3 (the aldehyde dehydrogenase 1 family, member A3) are associated with developmental defects. The ALDH1A3 enzyme catalyzes retinoic acid biosynthesis and is essential to patterning and neuronal differentiation in the development of embryonic nervous system. Several missense mutations in ALDH1A3 have been identified in family studies of autosomal recessive microphthalmia, autism spectrum disorder, and other neurological disorders. However, there has been no evidence from animal models that verify the functional consequence of missense mutations in ALDH1A3. Here, we introduced the equivalent of the ALDH1A3 C174Y variant into the Caenorhabditis elegans ortholog, alh-1, at the corresponding locus. Mutant animals with this missense mutation exhibited decreased fecundity by 50% compared to wild-type animals, indicating disrupted protein function. To our knowledge, this is the first ALDH1A3 C174Y missense model, which might be used to elucidate the effects of ALDH1A3 C174Y missense mutation in the retinoic acid signaling pathway during development.
Description
Retinoic acid (RA), the active metabolite of vitamin A, broadly regulates gene expression. The RA signaling pathway plays an essential role in embryonic development, including the development of the body axis, eye, brain, and heart (Ghyselinck & Duester, 2019). One of the key enzymes in the biosynthesis of RA is aldehyde dehydrogenase 1 family member A3 (ALDH1A3). ALDH1A3 converts retinaldehyde to retinoic acid and it is expressed early in forebrain development (McCaffery & Drager, 1994). Several mutations in ALDH1A3 have been implicated in patients with autosomal recessive microphthalmia and other neurological disorders (Fares-Taie et al., 2013; Roos et al., 2014). However, current animal models of ALDH1A3 have large truncations of the protein. Direct evidence of the effects of missense variants on ALDH1A3 protein activity has not yet been obtained.
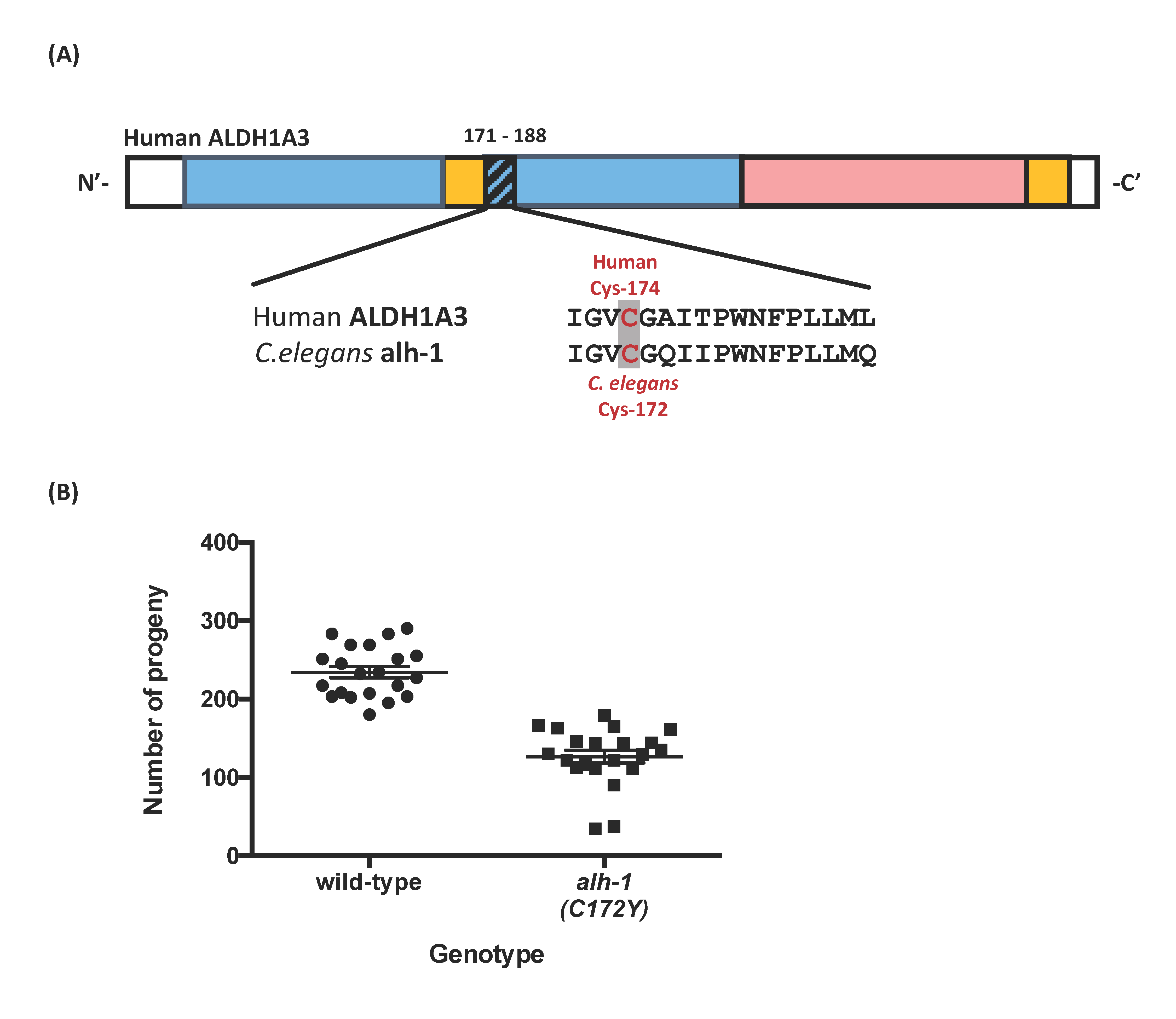
In this study, we aimed to determine the functional consequence of ALDH1A3(C174Y) missense variants implicated in patients (Roos et al., 2014). Patients with the Cys174Tyr missense variant presented with autosomal recessive microphthalmia, autistic symptoms, and intellectual disabilities. The human ALDH1A3 C174Y missense residue is located at the amino-terminal nicotinamide adenine dinucleotide (NAD)-binding domain (Moretti et al., 2016), which is evolutionarily conserved between human and commonly used model organisms, such as C. elegans, Drosophila, zebrafish, and mouse. The ALDH1A3 orthologs in these model organisms have similar amino acids at >75% of positions of the human ALDH1A3 protein. We believe the conservation of protein sequences among various species indicates the essential role of this domain in protein functions. Here, we use C. elegans as a prime candidate for studying the causality of missense variants in ALDH1A3. We generated the targeted ALDH1A3 C174Y missense variant in its C. elegans orthologous gene alh-1 (Figure 1A).
The short lifespan and genetically modifiable nature of C. elegans allow rapid screening of functional impactful missense mutation implicated in human diseases. We previously established a working pipeline to introduce autism-associated missense mutations into the genome of C. elegans using CRISPR/Cas9 and homology-directed repair (Wong et al., 2019). Here, we used this pipeline to generate a missense mutant, alh-1 (C172Y), that matches the human ALDH1A3(C174Y) variant. We then characterized this C. elegans missense mutant in a fecundity assay to quantify the robustness of germline development. The C. elegans germline development is a highly sensitive process, which reflects subtle changes in sensory perception, feeding behavior, and metabolism (Hubbard et al., 2013). As a result, we used the C. elegans fecundity assay as a screening tool to identify functional changing missense mutations, as defects in germline development will result in a decreased brood size.
We evaluated the impact of C172Y missense mutation on development using a fecundity assay, which measures the number of viable progeny per hermaphrodite. The alh-1(C172Y) missense mutant nematode, alh-1(sy899), showed only 54% of wild-type fecundity (Figure 1B. Wild-type: 234 ± 7; alh-1(C172Y): 127 ± 8. p < 0.0001). We observed that two of the eighteen mutant animals had very low brood size and a lot of unhatched eggs (17 unhatched eggs to a brood size of 34, and 4 unhatched eggs to a brood size of 38). The other animals had essentially no unhatched eggs or dead larvae. This result indicated a malfunction in the aldehyde dehydrogenase protein, resulting in partial sterile and germline developmental defects. Our result is consistent with the observation that the C. elegans alh-1(tm5823) putative null mutant strain produces some dead animals and a highly expressed sterile phenotype (personal communication with Dr. Shohei Mitani, NBPJ). The alh-1(tm5823) mutant harbors a 618 bp deletion, located outside of our C172Y missense site and removes part of the NAD-binding and catalytic domains. Our conserved C172Y missense residue is also located in the NAD-binding domain. In addition, the developmental phenotype is displayed in other animal models of the aldehyde dehydrogenase family. For example, Aldh null larvae and adults are less viable in the presence of ethanol in Drosophila (Fry & Saweikis, 2006). Moreover, mouse Aldh3-/- homozygotes were impaired in early forebrain development, possibly through failure to induce genes needed to establish regionalization (Molotkova et al., 2007). The Aldh1a3 null mouse showed perinatal lethality that could be rescued by maternal RA treatment (Dupé et al., 2003).
The developmental defects caused by mutations in ALDH1A3 are likely to act through alteration in the RA signaling pathway. RA binds to its nuclear RA receptor (RAR), which forms a heterodimer with retinoid X receptors (RXRs) and RA response elements (RARE). The RAR/RXR/RARE complex regulates gene transcription at specific locations during various developmental stages. In addition, RA is known to promote the differentiation of neurons prenatally; RA concentration, indicated indirectly by ALDH1A3 expression, influences the maturation of selected parts of the cerebral cortex postnatally (Wagner et al., 2006). This could be a possible explanation of a connection between ALDH1A3 mutations and the neurological symptoms displayed in patients with intellectual disabilities and autism spectrum disorder. Our result illustrated that ALH-1(C172Y) residue is necessary for wild-type ALH-1 protein function in C. elegans. Since the ALH-1(C172Y) residue is conserved between C. elegans and humans, it is likely that this residue also has a functionally significant role in human ALDH1A3 protein. Further study in other model organisms is needed to validate this and potentially elucidate the mechanistic basis of the functional consequences of the ALDH1A3(C174Y) variation.
Methods
Request a detailed protocolFecundity assay
Well-fed C. elegans were synchronized at the L4 stage. In the fecundity assay, individual L4 hermaphrodites were placed on separate NGM plates seeded with OP50, and these animals were subsequently transferred to a new plate every day. The number of newly hatched larvae progeny was counted for every plate 1 day after the adult was transferred. The total fecundity comprises the sum of progeny produced for four days per animal.
Statistical analysis
The fecundity assay was analyzed by Mann-Whitney test using GraphPad Prism version 6 (GraphPad, La Jolla, CA).
Reagents
The Bristol N2 C. elegans strain was used as the wild-type control and background for the CRISPR experiments (Brenner, 1974). The alh-1(C172Y) missense strain, PS7481 alh-1(sy899), was generated using the protocol described previously (Wong et al., 2019). Detection primers sequence: forward primer 5’-TACAGTTATTACGCCGGATGG-3’; reverse primer 5’- CATGTGCGACGAAATAGCTTG -3’. The expected PCR product length for the wild-type is 370 bp; PCR product from mutant strain can be further digested by HaeIII (New England Biolabs, Ipswich, MA) into two fragments of 104 and 266 bp. All strains were maintained on nematode growth medium (NGM) agar plates seeded with Escherichia coli OP50 at room temperature (21 ± 1◦C).
Acknowledgments
This work was supported by Simons Foundation (SFARI award # 367560 to PWS). PWS was an investigator with the Howard Hughes Medical Institute during the initial part of this study.
References
Funding
Simons Foundation (SFARI award # 367560)
Reviewed By
AnonymousHistory
Received: December 29, 2020Revision received: January 8, 2021
Accepted: January 11, 2021
Published: January 14, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Wong, WR; Maher, S; Oh, JY; Brugman, KI; Gharib, S; Sternberg, PW (2021). Conserved missense variant in ALDH1A3 ortholog impairs fecundity in C. elegans. microPublication Biology. 10.17912/micropub.biology.000357.Download: RIS BibTeX
