
Abstract
Polymerase Chain Reaction (PCR) is a powerful tool to detect natural variation or experimentally introduced variation in research and clinical settings and a widely-used method for genotyping. Single nucleotide polymorphisms (SNP) detection is challenging by PCR as the variant and wild type alleles differ by only one nucleotide. Traditional methods to detect SNPs, including Sanger sequencing and commercial kits, are usually time-consuming. Here we describe a simple primer design strategy that enables specific variant detection through regular one-step PCR. The strategy employs the differential efficiency of genomic PCR using a primer that has a single mismatch with the chromosome that contains the SNP to be detected (typically the variant allele) versus two mismatches with the corresponding alternative allele (typically the wild type allele). To date, we have successfully employed this approach to detect more than 20 SNPs. The simplicity and robustness of the approach allows rapid application to legacy mutations as well as newly discovered or generated SNPs.
Description
Single nucleotide polymorphisms (SNPs) are the most common genetic variation between natural populations of a species, are the frequent cause of phenotypes observed in organisms from forward genetic chemical mutagenesis screens, and are employed to probe the functional consequences of missense and noncoding changes made by CRISPR/Cas9 gene editing (Brenner, 1974; Okamoto et al., 2019; Thompson et al., 2013; Zhao et al., 2014). For model organisms, SNP genotyping is essential for strain construction and is an important part of strain validation necessary for research rigor and reproducibility. This is particularly the case if the SNP does not have a known phenotype, does not have an easy way to score phenotype, or the phenotype is masked by epistasis. Additionally, laboratories are typically working with multiple genes, often examining multiple alleles, which could be legacy mutations or newly discovered/generated variants. Therefore, a simple SNP detection approach is desirable to allow rapid genotyping. Furthermore, the approach should be nimble in the sense of being applicable to any gene, potentially any SNP, and the generation of a new assay and its execution being straightforward and occurring in a short time frame. Here we describe a simple system for single worm genotyping. The strategy employs the differential efficiency of genomic PCR using a primer that has a single mismatch with the chromosome that contains the SNP to be detected (typically the variant allele) versus two mismatches with the corresponding alternative allele (typically the wild type allele). The genotype of a worm is deduced from two samples of the single worm lysis, where one PCR reaction uses a primer with mismatches to specifically detect the variant and not the wild type allele and the other PCR reaction uses a primer with mismatches to specifically detect the wild type allele and not the variant.
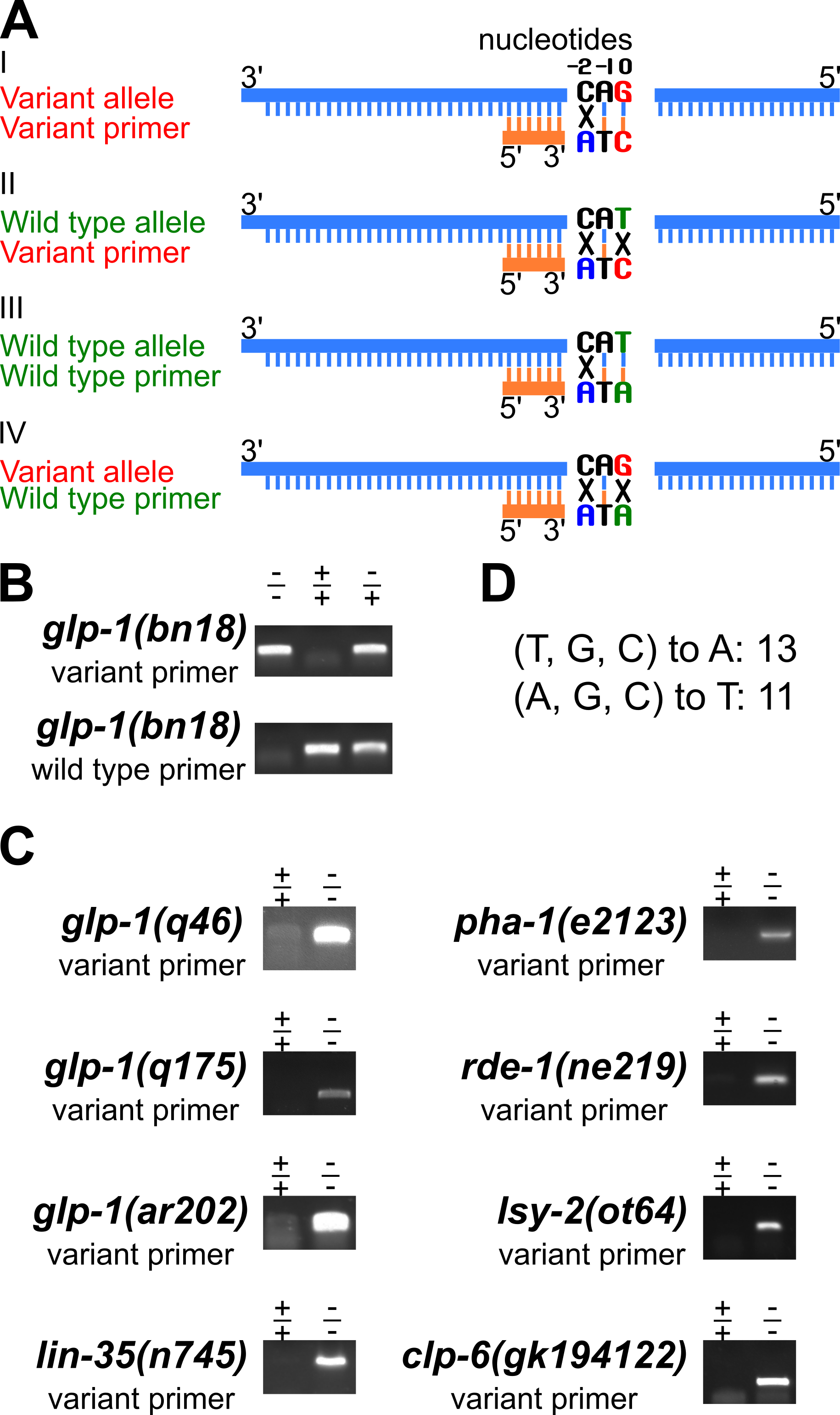
Key to the approach is a robust PCR method to detect one allele but not the other at a SNP residue (Figure 1A). A forward and a reverse primer are needed for each PCR reaction. We have designed the forward primer to more efficiently amplify one of the alleles. There are two important features of the forward primer. First, the 3’ nucleotide of the primer is at the SNP, denoted as 0 position, and pairs with the SNP residue to be detected but is mismatched with the other allele residue. Therefore the variant and wild type primers have different bases at their 3′ end. Second, two bases upstream of the SNP position, denoted as -2 position, an additional change was introduced that is a mismatch with both the variant and wild type alleles (Figure 1A, in blue in examples I to IV), to further discriminate between the two alleles in PCR. The reverse primer is identical in both cases. As shown in Figure 1A examples I to IV, the variant allele “G” and complement “C” are highlighted in red, and the wild type allele “T” and complement “A” are in green (Figure 1A). The variant primer has one mismatch relative to the variant allele (Fig. 1A, example I), compared to two mismatches relative to the wild type allele (Fig. 1A, example II). Therefore the variant primer can selectively amplify the variant allele. Similarly, the wild type primer can specifically detect the wild type allele (Figure 1A, examples III and IV). We have successfully applied this primer design strategy to detect the glp-1(bn18) variant allele (Kodoyianni et al., 1992) and the wild type allele through PCR (Figure 1B; genomic position III: 9098493, WormBase). By sampling the single worm lysis twice and performing separate PCR reactions with primers for the variant and the wild type allele, the genotype at the glp-1 bn18 residue is determined. As shown in Figure 1B, the variant primer preferentially amplifies the variant allele, with minimal background signal detected within wild type sample. Similarly, the wild primer preferentially amplifies the wild type allele, with minimal background signal detected within the homozygous bn18 variant sample. Gel images for an additional eight SNPs with variant specific primers are presented in Figure 1C. A summary of the nucleotide changes introduced at -2 position is shown in Figure 1D. To date, we have applied this approach to detect more than 20 SNPs, including additional glp-1 alleles (Kodoyianni et al., 1992; Pepper et al., 2003) and other genes (Johnston & Hobert, 2005; Lu & Horvitz, 1998; Mani & Fay, 2009; Tabara et al., 1999; Thompson et al., 2013). In summary, the primer design strategy presented here can be easily applied to legacy and newly identified SNPs, reduces time/effort and reagent costs, significantly simplifying SNP detection and genotyping by PCR in C. elegans, and likely in other systems.
Methods
Request a detailed protocolAll primers designed to have Tm between 58 °C to 61 °C. GoTaq DNA Polymerase (Promega, cat#M3008) was used in all PCR reactions described, following the manufacturer’s instructions. The single worm lysis PCR procedure is as previously described (Barstead et al., 1991; Williams et al., 1992). All PCR used the same cycling program. Initial denaturation of genomic DNA occurred at 94 °C for 2 minutes, 35 repeats of following: denature at 94 °C for 20 seconds, annealing at 55 °C for 20 seconds, and extension at 72 °C for 30 seconds. Final extension occurred at 72 °C for 2 minutes. No special treatment was required before the PCR samples were loaded on an agarose gel.
Reagents
Table 1: primers used in this study
| Primer_name | Primer_seq |
| 650_glp-1_bn18_variant_F | gatgaattggaccggaatggtatgaAtA |
| 649_glp-1_bn18_wildtype_F | gatgaattggaccggaatggtatgaAtG |
| 190_glp-1_bn18_R | agagctgttcgtcctttatacttgt |
| 20_glp-1_q46_variant_F | gggcaaagaccattctccaaatAtT |
| 21_glp-1_q46_R | ctccatcgcctcgtctttcaatac |
| 766_glp-1_q175_variant_F | ggaaaatccggtcgatattgtgTaT |
| 767_glp-1_q175_R | gcagtgtggtctctgtagtggaa |
| 630_glp-1_ar202_variant_F | cagggtattgacatttggagaatggtctttAcT |
| 260_glp-1_ar202_R | gagccacttggagtataatgacgatg |
| 674_lin-35_n745_variant_F | ccaaatgacattgttactggtgcaAgA |
| 675_lin-35_n745_R | tgtcaagcatttcagcaacgga |
| 684_pha-1_e2123_variant_F | taacttgatgaacatcggtaatcatacTgT |
| 685_pha-1_e2123_R | cttaatgcccttgcaccgtagt |
| 646_rde-1_ne219_variant_F | gtggcttctcatgaacttcaagatgAtT |
| 192_rde-1_ne219_R | aaatcggacagaggaagaaatgca |
| 692_lsy-2_ot64_variant_F | gatctgtgtgtatcactgcatgAtA |
| 693_lsy-2_ot64_R | ctgaagaagatgagatggtggaagg |
| 1149_clp-6_gk194122_variant_F | ggcagtcgatcatcaattactacatcaTcT |
| 1150_clp-6_gk194122_R | ccttgttgggtcatttccacgt |
Notes to the table 1: The bold nucleotide at 3’ end of the forward primer denotes either the variant allele or the wild type allele. The italicized nucleotide denotes mismatch change that was introduced two bases upstream of the SNP site. F forward primer; R reverse primer.
References
Funding
R01 GM-100756 to TS
Reviewed By
AnonymousHistory
Received: April 22, 2021Revision received: May 27, 2021
Accepted: May 28, 2021
Published: June 11, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Chen, J; Schedl, T (2021). A simple one-step PCR assay for SNP detection. microPublication Biology. 10.17912/micropub.biology.000399.Download: RIS BibTeX
