Department of Zoology, University of British Columbia, Vancouver, British Columbia, Canada

Abstract
We used whole-genome sequencing (WGS) data from a Caenorhabditis elegans strain homozygous for the reciprocal translocation hT2(I;III) to identify its breakpoints molecularly. The translocation structure is fairly straightforward, with only minor secondary rearrangement in addition to the primary breakpoints. The graphical representation below depicts the two hT2 half-translocations for ease of conceptualization.
Description
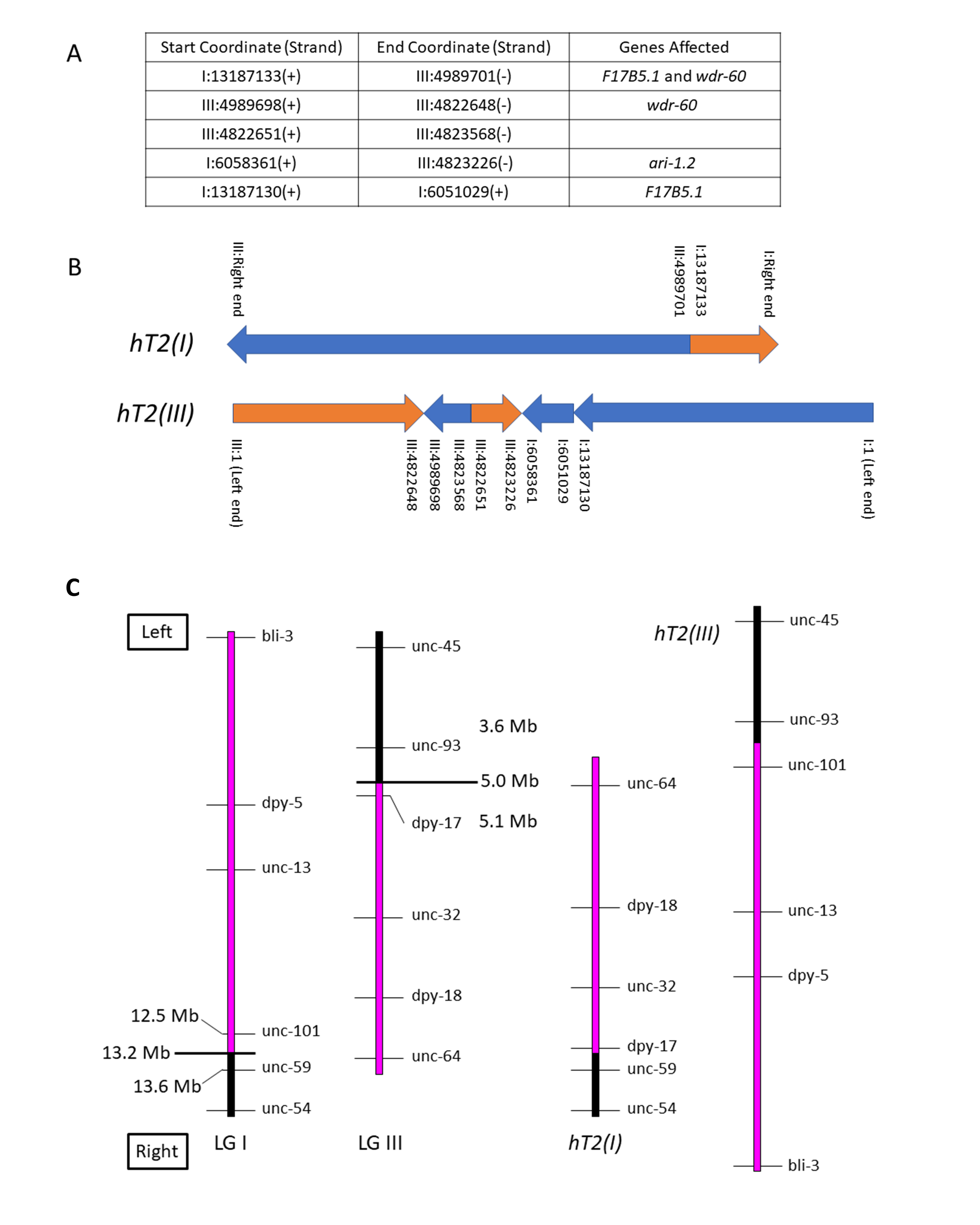
The reciprocal translocation hT2 was isolated after gamma irradiation of bli-4(e937)I males followed by crossing to unc-13(e450) I; dpy-18(e364) III hermaphrodites and screening F2 broods for evidence of pseudolinkage (McKim et al., 1993). Subsequent genetic experiments, including variants of hT2 with new morphological mutations induced by mutagenesis, elucidated its genetic structure as a reciprocal translocation and its meiotic pairing and segregation behavior. It is comprised of two half-translocations: hT2(I), which pairs with and disjoins from the normal LG I in hT2 heterozygotes; and hT2(III), which pairs with and disjoins from the normal LG III in heterozygotes. In simple terms, the mutagenesis event that produced hT2 cleaved LG I between unc-101 and unc-59, and LG III between dpy-17 and unc-93; in hT2, the resulting rightmost portion of LG III is translocated to the rightmost portion of LG I to form hT2(I), and the leftmost portion of LG I is translocated to the leftmost portion of LG III to form hT2(III) (see Figure 1).
The half-translocation hT2(I) runs from the right end of LG I through coordinate 13187133, where it joins to the right portion of LG III from coordinate 4989701 through the right end. The half-translocation hT2(III) runs from LG III coordinate 1 through coordinate 4822648, where it joins to a short complex rearrangement containing pieces of both LG I and LG III, and then to a portion of LG I from coordinate 1 through 13187130. These physical breakpoints correspond to the genetic limits of balancing behavior observed in recombination analysis of marker/hT2 trans heterozygotes. Such experiments showed that unc-101 (I: 12508299 – 12513420) is balanced, while unc-59 (I: 13629227 – 13633492) is not; and dpy-17 (III: 5107330 – 5108492) is balanced, while unc-93 (III: 3644375 – 3648822) is not.
hT2 has been used extensively as an effective balancer for the left portion of LG I from the left end through unc-101, and right portion of LG III from the right end through dpy-17 (see Figure 1). The original hT2 is homozygous viable and is marked with bli-4(e937)I, and several morphological variants exist (summarized in Edgley et al., 1995). The CGC maintains over 450 strains that utilize variations of this balancer for maintaining mutant alleles of essential genes.
Methods
Request a detailed protocolSequence Generation
A large asynchronous population of hT2[bli-4(e937)](I;III), established from a single Bli-4 segregant from VC386 (nmy-2(ok499)/hT2 I; +/hT2 [bli-4(e937)] III) was harvested by washing freshly-starved 100 mm standard agar/OP50 culture plates with M9 buffer. Worms were pelleted in 15 ml centrifuge tubes, the supernatant was removed by aspiration, and the packed worms were used to make purified DNA using standard extraction protocols. The sequencing library was made using Illumina’s Nextera XT library prep kit and run on an Agilent Bioanalyzer to check average fragment size. The sequencing was performed with Illumina MiSeq using 2 x 300 bp reads and an average coverage of 24X was acquired.
Sequence Analysis
The raw sequence data from this study have been deposited in the NCBI Sequence Read Archive (SRA; ncbi.nlm.nih.gov/sra) under accession number PRJNA780557. The sequencing reads were first aligned to the reference C. elegans genome version WS230 using the BWA aligner (Li and Durbin 2009). Read alignments were visually inspected in the regions of chromosomes I and III expected to contain the breakpoints associated with hT2 translocation using the IGV genome viewer (Thorvaldsdóttir et al., 2013). LUMPY (Layer et al., 2014) was also used to search for potentially missed breakpoints from the visual inspection. Each breakpoint was then further analyzed by aligning split reads at the breakpoint using the blast tool available on the WormBase website (version WS282, https://wormbase.org/tools/blast_blat), which allowed connecting two breakpoints to one another as reported in Fig. 1A. Mutated chromosomes I and III for hT2 were then deduced using those breakpoint connections and a parsimonious approach while taking into account the copy-number evidence provided by the read coverage in the regions of interest.
Reagents
DNA sequence analysis was based on the wild type N2 Bristol derived strain PD1074 and a Bli hT2 homozygous derivative of strain VC386 nmy-2(ok499)/hT2 I; +/hT2 [bli-4(e937)].
Acknowledgments
We thank Aric Daul and Ann Rougvie for providing the information on hT2 balanced strains at the CGC.
References
Funding
This work was supported by Canadian Institute of Health Research grant PJT-148549 (awarded to DGM). This work was also supported by an R24 National Institute of Health grant 5R240D023041 (awarded to Ann Rougvie, Paul Sternberg, Geraldine Seydoux and DGM).
Reviewed By
Kim McKimHistory
Received: November 24, 2021Revision received: December 6, 2021
Accepted: December 7, 2021
Published: December 9, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Flibotte, S; Edgley, M; Au, V; Moerman, DG (2021). Whole Genome Sequencing identifies reciprocal translocation hT2(I;III) breakpoints.. microPublication Biology. 10.17912/micropub.biology.000505.Download: RIS BibTeX
