Division of Reproductive Sciences, Department of Obstetrics and Gynecology, Duke University Medical Center, Durham, NC
Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC
University of North Carolina at Chapel Hill, Chapel Hill, NC
Department of Biology, North Carolina State University, Raleigh, NC
Abstract
Maternal exposure to tobacco smoke during pregnancy has been associated with many negative child health outcomes. Tobacco smoke exposure alters DNA methylation in the developing embryo/fetus and may be a mechanism that increases risk of later life disease. Previous studies have identified CpG sites in umbilical cord blood that are associated with in utero tobacco smoke exposure. We sought to validate findings for CpG sites within several of the top hit genes, AHRR, CYP1A1, and GFI1, using targeted quantitative bisulfite pyrosequencing. Comparing results from cord blood specimens of tobacco smoke-exposed to unexposed newborns, we confirmed significance at all previously identified CpG sites tested, including one in AHRR (p=0.007), three in CYP1A1 (p<0.0001), and one in GFI1 (p=0.008). These assays also captured novel differentially methylated CpGs located near the identified sites that were not included in the prior array-based studies (p value range, 0.02 to <0.0001). These results validate the prior findings and provide a simplified and more economical approach to analysis of CpG sites for expanded use as biomarkers of in utero tobacco smoke exposure.
Description
The Developmental Origins of Health and Disease (DOHaD) hypothesis is based on anecdotal and empirical findings that early life exposures are associated with later life susceptibility to disorders and diseases (Barker 1990). Our susceptibility to in utero toxicant exposures is now a major focal point in public health research. Gestational exposure to tobacco smoke is associated with low birth weight (Sbrana et al. 2011), airway hyperreactivity associated with asthma (Lee et al. 2015), and neurodevelopmental effects such as attention deficit hyperactivity disorder (Nomura et al. 2010; Sciberras et al.2011). These outcomes have been linked to epigenetic alterations, including changes in DNA methylation (Suter et al. 2013; Markunas et al. 2014; Richmond et al. 2015).
A previous study utilizing the Infinium HumanMethylation450 (450k) BeadChip reported differential methylation related to in utero tobacco smoke exposure (Joubert et al. 2012) and identified 26 significant CpG probes. A number of these are in coding regions of the growth factor independent 1 transcription repressor (GFI1) and the aryl-hydrocarbon receptor repressor (AHRR) genes, in addition to a region upstream of the cytochrome P450 gene, CYP1A1. Further studies have validated these results using the same or similar array-based methods (Richmond et al. 2015; Joubert et al. 2016; Rotroff et al. 2016; Rzehak et al. 2016). Here we sought to validate these findings using bisulfite pyrosequencing assays to measure DNA methylation in umbilical cord blood leukocytes from infants who were unexposed or exposed to tobacco smoke during pregnancy.
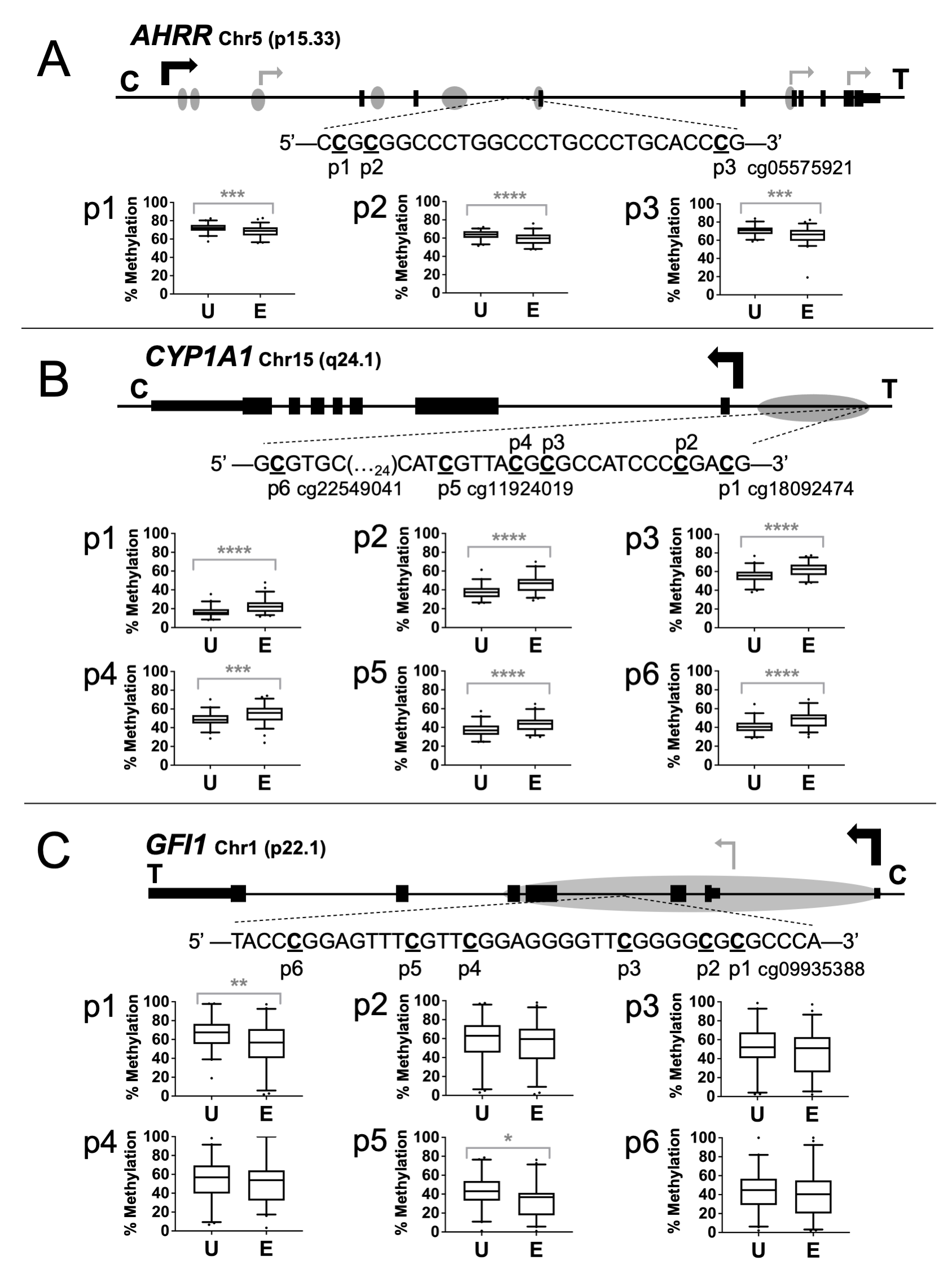
The sequence and positions of the specific CpGs analyzed are presented in Figure 1A-1C. The 146 bp PCR amplicon for AHRR contains three CpGs, one of which corresponds to probe cg05575921. The 226 bp CYP1A1 amplicon contains six CpG sites, including CpGs corresponding to probes cg22549041, cg11924019, and cg18092474. The 61 bp GFI1 amplicon is 61 bp long and contains six CpG sites, one of which corresponds to probe cg09935388.
Pyrosequencing assays were tested in duplicate or triplicate with mixtures of Qiagen’s Epitect fully methylated and unmethylated human DNAs containing 0%, 25%, 50%, 75% or 100% methylated DNA. Measured methylation was plotted against expected methylation levels. Goodness of fit assays for each CpG showed the assays measured incremental increases in DNA methylation as the amount of input methylated DNA increased across the range of possible values (AHRR: R2=0.97, p=0.0028; CYP1A1: R2 >0.96, p=0.0029; GFI1: R2>0.92, p=0.003).
The assays were then used to measure methylation in human umbilical cord blood DNAs from the Newborn Epigenetics STudy (NEST) cohort. Differential methylation was substantiated for the HumanMethylation450 probe CpGs for AHRR cg05575921 (p=0.0007) (Figure 1A), CYP1A1 cg11924019, cg22549041, and cg18092474 (p<0.0001) (Figure 1B), and GFI1 cg09935388 (p=0.008) (Figure 1C).
An advantage of targeted analyses by pyrosequencing is the ability to assess adjacent CpG sites that may not be represented on the 450k platform. For AHRR, sites p1 and p2 showed an average decrease in the exposed group of 3.9% (p=0.0009) and 4.9% (p<0.0001), respectively. For CYP1A1, sites p2, p3, and p4 showed an average increase in the exposed newborns of 8.4% (p<0.0001), 6.7% (p<0.0001), and 6.0% (p=0.0006), respectively. The GFI1 assay identified only one additional CpG site as significantly differentially methylated. Site p5 exhibited a 9.1% (p<0.02) decrease in the exposed group.
Using an independent quantitative method, we corroborated the findings from previously published studies of cord blood showing altered DNA methylation in association with in utero tobacco smoke exposure using array-based platforms (Joubert et al. 2012; Joehanes et al. 2016; Joubert et al. 2016; Tehranifar et al. 2018). We found significantly different levels of methylation between unexposed and exposed infant cord blood at identified sites for AHRR, CYP1A1, and GFI1. We also showed significant differences in methylation at adjacent CpG sites that were not represented on the arrays used in the prior studies. These CpGs may collectively be useful as biomarkers for in utero tobacco smoke exposure using a relatively cost-efficient and scalable platform.
AHRR and CYP1A1 are involved with the aryl-hydrocarbon receptor (AhR) pathway, which is known to facilitate changes in gene expression after toxicant exposure. Specifically, this pathway is known to mediate toxicity of polycyclic aromatic hydrocarbons (PAHs), several of which are found in cigarette smoke (Nguyen and Bradfield 2008). Although GFI1 has not been previously functionally associated with tobacco smoke exposure, it is involved with fundamental development processes such as hematopoiesis, inner ear and pulmonary neuroendocrine cell development, cellular proliferation, differentiation, and apoptosis, and pre-mRNA splicing control as well as immune system response (Duan et al. 2005; Khandanpour et al. 2011; Moroy and Khandanpour 2011; Joubert et al. 2012; Rotroff et al. 2016).
A limitation of the study was that we used self-reported exposure rather than cotinine levels for some cases in which these data were missing. There is a tendency for participants to underestimate exposure, which is either intentional under-reporting due to societal expectations or a lack of awareness of secondhand exposure. The overall effect of under-reporting would diminish the differences between groups, as the group classified as unexposed may contain exposed individuals.
In conclusion, we have shown concordance between the previously reported 450k BeadChip findings and those from bisulfite pyrosequencing of an independent cohort. Our results provide the foundation for implementation of these relatively low-cost assays for defined analysis of these regions in other human cohort studies seeking to define exposure effects on these vulnerable regions of the genome.
Methods
Request a detailed protocolSamples. This research was approved by the Duke Institutional Review Board (protocol Pro00043033). Cord blood specimens were selected based on self-reported smoking status during pregnancy (exposed, mother or other person living in the household smoked tobacco during her pregnancy; unexposed, mother did not smoke and was not exposed to tobacco smoke in her residence during pregnancy). These specimens were derived from the Durham, NC-based Newborn Epigenetics STudy (NEST), a mother-infant prospective cohort designed to determine the influence of early life exposures on the epigenome and health-related outcomes [see (Hoyo et al. 2014)].
Pregnant women were recruited from 2005 to 2011. Cord blood DNA was extracted from leukocytes separated by centrifugation from whole blood collected in vacutainer tubes containing EDTA. Samples were included in the analyses if we obtained clean PCR amplicons, the numbers of which were as follows: AHRR—42 unexposed, 58 exposed; CYP1A1—43 unexposed, 59 exposed; GFI1—41 unexposed, 50 exposed. Of these, maternal blood collected at enrollment for ~88% underwent measurement for cotinine, a major metabolic breakdown product of nicotine. For 12 women with missing cotinine values, self-report of tobacco exposure was used for exposure status classification. There was 87% congruence between self-reported exposure and exposure status from cotinine levels for the 820 women with plasma cotinine measurements in the parent study (Schechter et al. 2018).
DNA Methylation Analysis. Genomic DNA was purified using Puregene Reagents (Qiagen) and 800 ng was modified with sodium bisulfite using the Zymo EZ DNA Methylation kit (Zymo Research). Pyrosequencing assays were designed to determine methylation levels at the previously identified significant CpG sites in AHRR, CYP1A1, and GFI1 14. All assay designs included adjacent CpGs not included on the array platform used in prior studies. Primers (Sigma) were designed using Qiagen Assay Design Software 1.0.6. AHRR PCR primers: F, 5’-TGG GGA TTG TTT ATT TTT GAG AG-3’; R, 5’-[biotin]AAA AAA CCC TAC CAA AAC CAC TC-3’. CYP1A1 PCR primers: F, 5’-[biotin]ATG GGA GGT GAG GGG ATT-3’; R, 5’-CCC CAA TAC CAT TTA ACA TAA C-3’. GFI1 PCR primers: F, 5’-AGG GAG TTA AGT GGT TAG ATA A-3’; R, 5’-[biotin] TAT ACC CAC AAC ACT CCA ATT C -3’. PCR was performed in 8-well strip tubes with total reaction volumes of 10 µL per well: 1 µL (~20 ng) BS-treated DNA and 9 µl master mix containing 5 µL PyroMark PCR buffer, 0.6 µL 25mM MgCl2, 1 µL 10x Coral Load (Qiagen), 0.12 µL of both primers at 10 µM (Sigma), and 2.16 µL molecular grade water. PCR conditions were: AHRR, 95°C for 15 minutes; then 55 cycles of 94°C for 30s, 58°C for 30s and 72°C for 30s; final extension 72°C for 10 minutes; CYP1A1, 95°C for 15 minutes; then 55 cycles of 94°C for 30s, 55°C for 30s and 72°C for 30s; final extension of 72°C for 10 minutes; GFI1: 95°C for 15 minutes; then 5 cycles of 95°C for 30s, 66°C for 30s and 72°C for 30s; 5 cycles of 95°C for 30s, 63°C for 30s and 72°C for 30s; 5 cycles of 95°C for 30s, 60°C for 30s and 72°C for 30s; then 45 cycles for of 95°C for 30s, 57°C for 30s, and 72°C for 30s with final extension of 72°C for 10 minutes.
Sequencing primers were: AHRR, 5’-TTG TTT ATT TTT GAG AGG GTA-3’; CYP1A1, 5’- CCA AAA AAA AAA AAA TTA TAT T-3’; and GFI1: 5’-TTA AGT GGT TAG ATA AGG AT-3’. Validation of pyrosequencing assays was performed using EpiTect DNAs (Qiagen). Pyrosequencing was performed on a Qiagen Pyromark Q96 MD instrument and percent methylation was determined using Qiagen PyroMark CpG software 1.0.11.
Statistical analysis. All statistical analysis was conducted using GraphPad Prism version 7.03. Data from the unexposed and exposed samples were tested for normal distribution using the D’Agostino and Pearson normality test, and statistical differences between the two groups were determined using the Welch’s t-test for normally distributed data or Mann-Whitney test for data with a non-normal distribution.
Acknowledgments
We thank the Newborn Epigenetics STudy participants without whom this research would not be possible.
References
Funding
This work was supported by NIEHS P01ES022831, USEPA RD-83543701, NCRR UL1TR001117 and NIEHS T32-ES021432.
Reviewed By
Wilfried KarmausHistory
Received: August 23, 2021Revision received: December 5, 2021
Accepted: December 29, 2021
Published: January 7, 2022
Copyright
© 2022 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Crute, C; Liao, Y; Son, E; Grenier, C; Huang, Z; Hoyo, C; Murphy, SK (2022). Validation of differential DNA methylation in newborns exposed to tobacco smoke during gestation using bisulfite pyrosequencing. microPublication Biology. 10.17912/micropub.biology.000509.Download: RIS BibTeX
