
Abstract
Camptothecin (CPT) is a specific inhibitor of the DNA topoisomerase I (Top1p), currently used in cancer therapy, which induces DNA damage and cell death. Top1p is highly active at the repeated ribosomal DNA locus (rDNA) to relax DNA supercoiling caused by elevated transcription and replication occurring in opposite directions. Fob1p interacts with, and stabilizes, Top1p at the rDNA Replication Fork Barrier (rRFB), where replication and transcription converge. Here, we have investigated if the absence of Fob1p and the consequent loss of Top1p specific targeting to the rRFB impact the sensitivity and the cell cycle progression of wild-type cells to CPT. We have also investigated the consequences of the absence of Fob1p in rad52∆ mutants, which are affected in the repair of CPT-induced DNA damage by homologous recombination. The results show that CPT sensitivity and the global cell cycle progression in cells exposed to CPT is not changed in the absence of Fob1p. Moreover, we have observed in fob1∆ cells treated with CPT that the homologous recombination factor Rad52p still congregates in the shape of foci in the nucleolus, which hosts the rDNA. This suggests that, in the absence of Fob1p, Top1p is still recruited to the rDNA, presumably at sequences other than the rRFB, and its inhibition by CPT leads to recombination events.
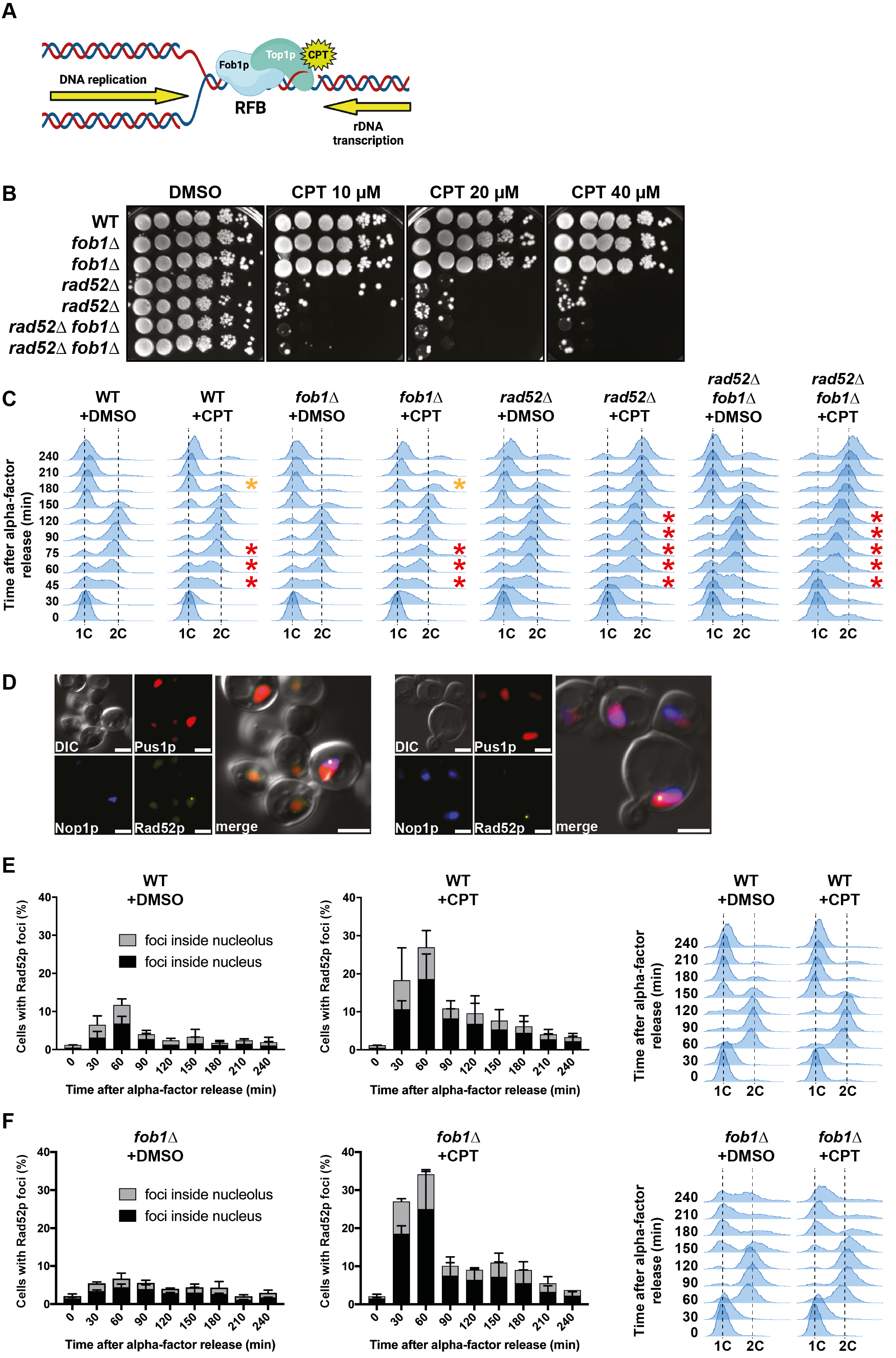
Description
DNA topoisomerases play a vital role in solving topological constrains during DNA replication and transcription (Pommier et al. 2016). Top1p is a DNA topoisomerase that relaxes DNA supercoiling by nicking the DNA, creating a covalent bond between the enzyme and the 3’ end of the DNA, called Top1p cleavage complex (Top1cc). Once the DNA is relaxed, Top1p religates the break by reversing its covalent binding. Top1p activity can be inhibited by drugs such as camptothecin (CPT), whose derivatives (topotecan, irinotecan) are largely used in cancer therapy. CPT binds to the Top1cc and delay the religation reaction, thus blocking Top1p on DNA (Figure 1A) (Pommier et al. 2006). It has been shown that treatment of cells with CPT induces DNA double-strand breaks (DSBs) specifically during DNA replication. One model proposes that the DNA nick is converted into a DSB by the passage of the replication fork, which separates the parental duplex DNA (Strumberg et al. 2000). Because CPT prevents Top1p ability to remove topological stress (Koster et al. 2007), another model proposed that the accumulation of topological constrains would block replication fork advance and induce DSB formation as a consequence of fork cleavage by the MUS81 nuclease (Regairaz et al. 2011). These DSBs must be repaired, otherwise mutations could arise and affect genome stability. DSBs are mainly repaired by Homologous Recombination (HR) during the S and G2 phases of the cell cycle, the Rad52p protein being a key player in this pathway in yeast Saccharomyces cerevisiae (Pardo et al. 2009). Rad52p proteins relocalize into discrete sub nuclear foci upon DSB formation (Lisby et al. 2003). In response to CPT treatment, yeast cells accumulate Rad52p foci both in the nucleus and the nucleolus (Stuckey et al. 2015). These results suggest that CPT-induced DSBs arise in the ribosomal DNA (rDNA) genes, which reside in the nucleolus. These genes are organized on the chromosome XII in a single cluster of 150-200 tandem repeats (Kobayashi 2011). Each rDNA repeat includes a Replication Fork Barrier (rRFB) bound by Fob1p, whose function is to avoid collisions between replication and transcription machineries by stalling the movement of replication forks in only one direction (Kobayashi and Horiuchi 1996). In the rDNA, it has been shown that Top1ccs accumulate naturally and specifically at the rRFB, and that this accumulation is completely lost in the absence of Fob1p (Krawczyk et al. 2014). DSBs have also been detected at the rRFB and are dependent on Top1p and Fob1p. These DSB signals were increased upon the inhibition of Top1p by CPT (Krawczyk et al. 2014), suggesting that the accumulation of Fob1p-dependent Top1ccs at the rRFB leads to DSB formation. Since the rDNA represents about 10% of the genome size of Saccharomyces cerevisiae and these DSBs could be detrimental for cell survival, we thus investigated if the absence of Fob1p could improve the resistance of yeast cells to CPT.
In order to test if FOB1 deletion could improve cell resistance to CPT, we combined the rad52∆ mutation, which confers hypersensitivity to CPT, to the fob1Δ mutation. fob1∆ cells did not show sensitivity to CPT at all tested concentrations (Figure 1B). Moreover, the hypersensitivity of rad52Δ cells was not alleviated by fob1Δ (Figure 1B). These results suggest that the presence of Fob1p and the consequent stabilization of Top1ccs at the rRFB is not the main cause of cell death in the absence of Rad52p when exposed to CPT.
We then analyzed the progression of cells by flow cytometry during a single cell cycle in response to CPT exposure. After the synchronization in G1 (1C DNA content), cells were incubated with either DMSO (control) or CPT in a minimal medium increasing cell permeability to CPT (Pardo et al., 2020) and released into S phase. WT cells treated with DMSO entered the S phase at 30 minutes and reached the G2 phase (2C DNA content) at 90 minutes. At 150 minutes, cells finished mitosis and progressed to the following G1, where they remained arrested by a second addition of the synchronizing agent α-factor (Figure 1C). WT cells exposed to CPT progressed slower through S phase and reached the G2 phase 30 minutes later than control cells. Consequently, these cells reached the following G1 with a 30 minutes delay (Figure 1C). Cell cycle progression of fob1∆ cells was comparable to WT, either incubated with DMSO or CPT, respectively. On the contrary, rad52∆ cells exposed to CPT showed an increased S-phase delay compared to WT (Figure 1C), consistent with a previous observation (Pardo et al. 2020). Once they reached the G2 phase 150 minutes after the release from the G1 synchronization, they remained blocked in G2/M until the end of the time course experiment, at 240 minutes (Figure 1C). These results confirm that Rad52p is important for cells to progress through S phase and complete mitosis when they are exposed to CPT (Pardo et al. 2020). FOB1 gene deletion did not change the cell cycle progression of rad52∆ cells, whether treated with DMSO or with CPT (Figure 1C). These data indicate that the absence of Fob1p does not impact the global cell cycle progression in cells exposed to CPT.
Finally, we used a Rad52 protein tagged with YFP to follow by fluorescence microscopy the formation of Rad52p-YFP foci in response to CPT exposure in WT and fob1∆ cells. After G1 synchronization, cells were released into S phase with DMSO/CPT. In WT cells treated with DMSO, Rad52p foci formed specifically during S-phase (30-60 min after release) and decreased until mitosis completion (Figure 1E). The foci were localized both in the nucleolus (marked by Nop1p-CFP) and in the remaining of the nucleus (marked by mCherry-Pus1p) (Figure 1D-E). The percentage of WT cells containing Rad52p-YFP foci increased about 3-fold in response to CPT and followed the same kinetics as control cells (Figure 1E). fob1∆ cells showed similar results (Figure 1F), suggesting that DNA lesions, presumably DSBs, still form in the rDNA in the absence of Fob1p.
Altogether, these results show that FOB1 deletion does not suppress the phenotypes induced by CPT exposure in WT or rad52∆ cells. The fact that Rad52p foci formation increased upon CPT exposure (Figure 1E) suggests that Rad52p foci are related to Top1ccs. Rad52p foci still formed in the nucleolus in fob1∆ cells exposed to CPT (Figure 1F), suggesting that Top1ccs can still accumulate in the rDNA, likely at sequences other than the rRFB, in the absence of Fob1p. This is consistent with the low amount of Top1ccs detected in the 35S gene, located upstream of the rRFB, in the presence or absence of Fob1p without CPT treatment (Krawczyk et al. 2014). Even if present in a lower amount, these Top1ccs further stabilized by CPT do not impact the cell cycle progression, nor the cell viability in fob1∆ cells (Figure 1B-C). One explanation could be that they do not give rise to DSBs, which are lethal DNA lesions. Indeed, the DNA fragments detected in the rDNA and interpreted as being the consequence of DSBs (Fritsch et al. 2010; Krawczyk et al. 2014; Sasaki and Kobayashi 2017) could also correspond to the reversal of replication forks (Kara et al. 2021), an event in which the fork goes backward by annealing the newly-synthetized DNA strands together. This hypothesis is supported by the lack of “DSB” signals accumulation at the rRFB in the absence of DSB repair factors (Fritsch et al. 2010; Sasaki and Kobayashi 2017; Kara et al. 2021) and by the fact that fork reversal events are increased in cells exposed to CPT (Ray Chaudhuri et al. 2012; Menin et al. 2018). Then, Rad52p foci formation could originate from recombination events initiated from the tip of reversed forks instead of DSBs.
Methods
Request a detailed protocolSensitivity assay. Yeast cells freshly grown on rich YPAD plates overnight at 30°C were resuspended in water and cell concentration was measured with a CASY flow cytometry apparatus. Cell concentration was normalized to 2×108 cells/ml. Tenfold serial dilutions we done and 5µl drops of each dilution were deposited on solid rich YPAD medium containing DMSO or CPT at different concentrations. Image were taken after 2 days of growth at 30°C. The experiment was repeated three times.
Flow cytometry. Exponentially growing cells at 7×106 cells/ml were synchronized in G1 by the addition of α-factor at 1 µg/ml for 2 hours at 30°C in rich YPAD medium. After cell synchronization in G1, cells were pelleted and resuspended in minimal MPD +SDS medium (0.17% yeast nitrogen base, 0.1% L-Proline, 2% glucose and 0.003% SDS) and incubated for 1h at 30°C in the presence of DMSO (control) or 100 µM CPT and α-factor at 1 µg/ml. Release into S phase was done by removing the α-factor by filtration and resuspending the cells in minimal MPD +SDS medium with DMSO or CPT. α-factor was again added 90 minutes after release. At the indicated time points, cells were recovered and fixed in ethanol 70%, permeabilized in Na-citrate 50 mM buffer, incubated with RNase A 50 µg/ml for 2 hours at 50°C and incubated with proteinase K (PK) 230 µg/ml for 1 hour at 50°C. DNA was stained with propidium iodide at 4 µg/ml for 2 hours at room temperature in the dark. DNA fluorescence was measured using Miltenyi Biotec MACSQuant analyzer and analyzed using FlowJo software. The experiment was repeated twice.
Microscopy. Exponentially growing cells at 7×106 cells/ml cultured in minimal SC medium without leucine were pelleted and resuspended in rich YPAD medium for synchronization in G1 by the addition of α-factor at 8µg/ml for 2.5 hours at 25°C. α-factor was removed by filtration and cells were released into S phase in rich YPAD medium containing DMSO (control) or 100 µM CPT. α-factor was again added 90 minutes after release. Cells were collected at the indicated time points and pictures of living cells were captured by fluorescence microscopy (×67) using a Zeiss Axioimager and ZEN software. Images were analyzed using ImageJ software and results were plotted using GraphPad Prism software. The experiment was repeated three times.
Reagents
Yeast strains used in this study come from the W303 background corrected for the rad5-535 mutation. Mutant strains were generated by PCR-mediated deletion and genetic crossing using standard methods. The strains used in Figure 1B-C (WT PP3241, fob1∆NAT PP3277 PP278, rad52∆klLEU2 PP3279 PP3280, rad52 ∆klLEU2 fob1∆NAT PP3281 PP3282) contain the bar1∆ mutation (without any marker) to facilitate the cell cycle synchronization in G1 phase. The strains used for microscopy in Figure 1D-F (WT PP4669 PP4670 and fob1∆NAT PP4672 PP4673) contain wild-type BAR1 gene, RAD52-YFP and mCherry-PUS1::URA3 integrated at their endogenous loci and the monocopy centromeric plasmid NOP1-CFP::LEU2. Alpha-factor was bought from BIOTEM company (custom synthesis). CPT stock solution was made from (S)-(+)-camptothecin (SIGMA C9911) dissolved in DMSO (SIGMA D8418) at 15 mM.
Acknowledgments
We thank María Moriel-Carretero for critical reading of the manuscript. We thank Danesh Moazed for the gift of the NOP1-CFP::LEU2 plasmid and Symeon Siniossoglou for the gift of the mCherry-PUS1::URA3 integrative plasmid. We acknowledge the MRI imaging facility, a member of the national infrastructure France-BioImaging, supported by the French National Research Agency (ANR-10- INBS-04, Investissements d’avenir).
References
Funding
This research was funded by a grant from the MSDAvenir fund (GnoStiC) to Philippe Pasero and by a grant from la Fondation ARC pour la Recherche sur le Cancer (ARCPJA22020060002119) to Benjamin Pardo.
Reviewed By
AnonymousHistory
Received: December 17, 2021Revision received: January 12, 2022
Accepted: January 13, 2022
Published: January 19, 2022
Copyright
© 2022 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Pourali, P; Pasero, P; Pardo, B (2022). Top1p targeting by Fob1p at the ribosomal Replication Fork Barrier does not account for camptothecin sensitivity in Saccharomyces cerevisiae cells. microPublication Biology. 10.17912/micropub.biology.000514.Download: RIS BibTeX
