Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA
Department of Pediatrics, Sanford School of Medicine, University of South Dakota, Sioux Falls, SD 57105 USA

Abstract
Molybdenum cofactor (Moco) is an essential prosthetic group that mediates the activity of 4 animal oxidases and is required for viability. Humans with mutations in the genes encoding Moco-biosynthetic enzymes suffer from Moco deficiency, a neonatal lethal inborn error of metabolism. Caenorhabditis elegans has recently emerged as a useful and tractable genetic discovery engine for Moco biology. Here, we identify and characterize K10D2.7/moc-6, the C. elegans ortholog of human MOCS2A, a sulfur-carrier protein essential for Moco synthesis. Using CRISPR/Cas9 gene editing, we generate 3 null mutations in K10D2.7/moc-6 and with these alleles genetically demonstrate that K10D2.7/moc-6 is necessary for endogenous Moco synthesis in C. elegans.
Description
The nematode C. elegans has recently emerged as a tractable system for genetic discovery in Moco biology (Warnhoff and Ruvkun 2019; Warnhoff et al. 2021). In C. elegans, endogenous Moco biosynthesis is not required for growth, development, and reproduction. C. elegans mutants defective in Moco biosynthesis are viable because they acquire and use Moco from their bacterial diet (Warnhoff and Ruvkun 2019; Warnhoff et al. 2021). Thus, C. elegans has 2 redundant pathways for maintaining cellular Moco: endogenous synthesis and dietary acquisition. C. elegans is the only known animal for which Moco biosynthesis is dispensable.
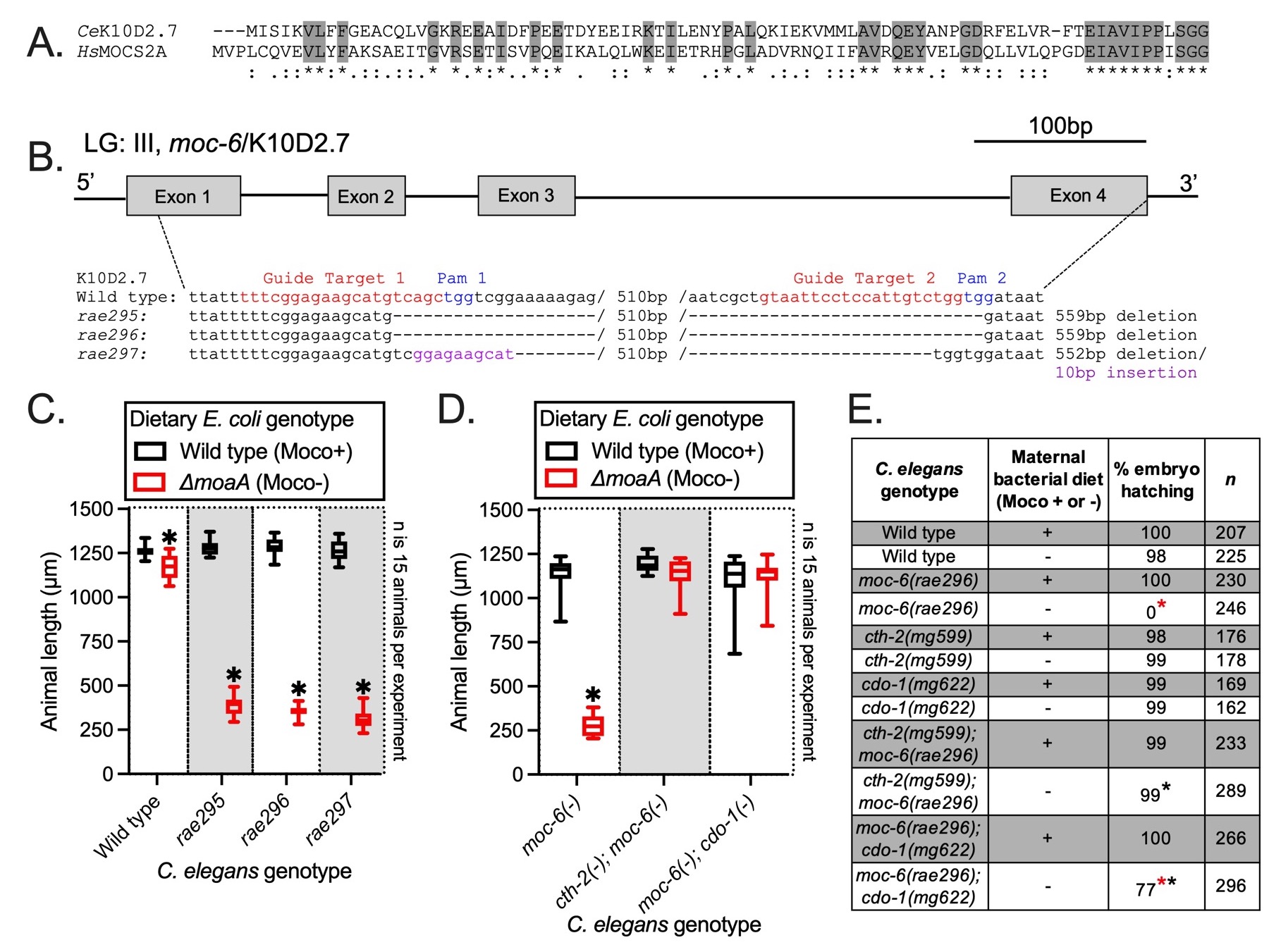
The human MOCS2A enzyme is required for Moco synthesis (Schwarz et al. 2009). MOCS2A is a small sulfur-carrier protein that is necessary for the conversion of cyclic pyranopterin monophosphate to molybdopterin. This reaction is the second step in the linear synthesis pathway of Moco from guanosine triphosphate (Schwarz et al. 2009). Loss-of-function mutations in MOCS2A cause human Moco deficiency, a rare and lethal inborn error of metabolism (Johnson et al. 2001). However, the C. elegans orthologue of MOCS2A has not been molecularly identified or genetically studied. Using amino acid sequence analysis, we determined that the C. elegans K10D2.7 genomic locus encodes a protein with high amino acid similarity and identity to human MOCS2A (Fig. 1A) (Altschul et al. 1990). To determine the role K10D2.7 plays in Moco biosynthesis, we used CRISPR/Cas9 to engineer 3 independent deletions of K10D2.7 (Fig. 1B) (Cho et al. 2013; Ghanta and Mello 2020). These K10D2.7 deletion alleles (rae295, rae296, and rae297) are likely null mutations because they eliminate >80% of the K10D2.7 coding sequence. As we have shown for other C. elegans mutations in Moco biosynthetic machinery, the K10D2.7 deletion alleles rae295, rae296, or rae297 cause no defects when animals are cultured under standard laboratory conditions feeding on wild-type E. coli which also synthesizes Moco. To determine if K10D2.7 is necessary for endogenous Moco synthesis, we cultured wild-type, rae295, rae296, and rae297 mutant animals on wild-type and ΔmoaA E. coli, a mutant bacterial strain that is unable to synthesize Moco. rae295, rae296, and rae297 mutant animals grew well on wild-type (Moco producing) E. coli but displayed a completely penetrant developmental arrest when cultured on ΔmoaA (Moco deficient) mutant E. coli (Fig. 1C). In contrast to K10D2.7 mutant animals, wild-type C. elegans grow well on wild-type or ΔmoaA mutant E. coli, although we observed a subtle, but statistically significant, reduction in the growth of wild-type animals on ΔmoaA mutant E. coli (Fig. 1C). By eliminating Moco from the diet, we revealed the defect in endogenous Moco synthesis caused by the rae295, rae296, and rae297 mutations. We conclude that K10D2.7 is required for C. elegans Moco synthesis and name this gene moc-6 (MOlybdenum Cofactor biosynthesis).
Moco deficiency in C. elegans also results in embryonic lethality when maternal animals are deprived of both endogenous and exogenous sources of Moco during reproductive adulthood (Warnhoff and Ruvkun 2019). moc-6 mutant C. elegans mothers were fed wild-type E. coli until the L4 stage of development, one stage prior to reproductive adulthood. These moc-6 mutant L4 mothers were then shifted onto wild-type or ΔmoaA mutant E. coli and the hatching rate of their progeny was determined. When moc-6 mutant animals were shifted to a Moco deficient diet, 0% of their embryos hatched (Fig. 1E). In contrast, 100% of moc-6 mutant embryos hatched when their mothers were fed wild-type E. coli. Hatching rates of wild-type embryos were not affected by the maternal diet (Fig. 1E). We conclude that maternal dietary Moco is sufficient to support the required Moco-dependent enzymes for embryonic development. Whether dietary Moco is acting maternally or embryonically to support embryonic development remains to be determined.
Previous genetic studies of the C. elegans Moco biosynthetic pathway demonstrated that Moco is required for the detoxification of sulfites produced by the sulfur amino acid metabolism pathway governed by cystathionase (cth-2) and cysteine dioxygenase (cdo-1). The activities of cth-2 and cdo-1 are required for the developmental arrest that occurs when C. elegans are defective in Moco synthesis and lack dietary Moco (Warnhoff and Ruvkun 2019). Loss-of-function/null mutations in cth-2 or cdo-1 suppress the developmental arrest displayed by moc mutant C. elegans grown on Moco-deficient E. coli (Warnhoff and Ruvkun 2019). To determine if the developmental arrest of moc-6 mutant C. elegans was also dependent upon cth-2 and cdo-1, we engineered cth-2; moc-6 and moc-6; cdo-1 double mutant animals and assessed their growth on wild-type and ΔmoaA mutant E. coli. In contrast to moc-6 single mutant animals, cth-2; moc-6 and moc-6; cdo-1 double mutant animals grew well on both wild-type and ΔmoaA mutant E. coli (Fig. 1D). cth-2 and cdo-1 single mutant animals grow well on both wild-type and ΔmoaA mutant E. coli (Warnhoff and Ruvkun 2019). Furthermore, mutations in cth-2 or cdo-1 suppressed the embryogenesis defects displayed by moc-6 mutant mothers fed on Moco-deficient E. coli (Fig. 1E). Although, we find that moc-6; cdo-1 double mutant embryos derived from mothers fed ΔmoaA mutant E. coli do not hatch at the same rate as wild type. Further studies are required to account for this observation. Together, these data demonstrate that cth-2 and cdo-1 inhibit normal development when Moco is deficient due to loss of both moc-6 and dietary Moco. CTH-2 and CDO-1 act in a catabolic pathway to produce sulfite from the sulfur amino acids methionine and cysteine. Sulfites are extremely toxic during Moco deficiency due to inactivity of the Moco-requiring sulfite oxidase enzyme, SUOX-1. Loss of cth-2 or cdo-1 prevents the metabolic production of sulfites, alleviates the cellular stress caused by inactive SUOX-1 (concomitant with Moco deficiency), and permits growth, development, and reproduction (Warnhoff and Ruvkun 2019). The mechanism by which sulfite promotes larval arrest and death remains enigmatic in both our C. elegans models and in human patients suffering from Moco deficiency.
This study demonstrates that moc-6 is required for endogenous C. elegans Moco biosynthesis and amino acid sequence analyses strongly suggest that moc-6 is orthologous to human MOCS2A.
Methods
Request a detailed protocolAnimal cultivation:
C. elegans strains were cultured using established protocols (Brenner 1974). Briefly, animals were cultured at 20°C on nematode growth media (NGM) seeded with wild-type E. coli (BW25113) unless otherwise noted. The wild-type C. elegans strain was Bristol N2. All C. elegans and E. coli strains used in this work are listed in the Reagents table.
Engineering deletions of moc-6/K10D2.7 using CRISPR/Cas9:
Genome engineering using CRISPR/Cas9 technology was performed using established techniques (Cho et al. 2013; Ghanta and Mello 2020). Briefly, 2 guide RNAs were designed and synthesized (IDT) that targeted the K10D2.7 locus (Fig. 1B). Cas9 (IDT) guide RNA ribonucleoprotein complexes were directly injected into the C. elegans germline (Cho et al. 2013). Newly induced deletions were identified in the offspring of injected animals using a PCR-based screening approach. The DNA primers used to screen for new deletions were: 5’-tggtgtagcggcaagtacaa-3’ and 5’-tccctattttgcaccctttg-3’. We were able to isolate and homozygoze 3 independent deletions of K10D2.7; rae295, rae296, and rae297 (Fig. 1B).
C. elegans growth assays:
C. elegans were synchronized at the first stage of larval development (L1). L1 animals were cultured on NGM seeded with wild-type or ΔmoaA mutant E. coli. Animals were cultured for 72 hours at 20°C and live animals were imaged using an SMZ25 stereomicroscope (Nikon) equipped with an ORCA-Flash4.0 camera (Hamamatsu). Images were captured using NIS-Elements software (Nikon) and processed using ImageJ. Animal length was measured from the tip of the head to the end of the tail. GraphPad Prism software was used to perform statistical tests as well as calculations of median, upper, and lower quartiles.
Quantification of embryo hatching:
To determine the hatching rate of wild-type and mutant C. elegans, we performed synchronized egg lays using young adult animals. Embryos were scored for hatching 12-24 hours after being laid. To test the effect of maternal dietary Moco on embryo hatching, mothers were initially grown on wild-type E. coli to the L4 stage of development and were shifted onto either wild-type or ΔmoaA mutant E. coli. Shifted L4 animals developed overnight into young adults on their respective E. coli diets and their progeny were assayed for their ability to hatch.
Reagents
| Organism | Strain | Genotype | Source |
| C. elegans | N2 | Wild type | CGC |
| C. elegans | USD954 | moc-6(rae295) III | This work |
| C. elegans | USD955 | moc-6(rae296) III | This work |
| C. elegans | USD956 | moc-6(rae297) III | This work |
| C. elegans | GR2257 | cth-2(mg599) II | CGC |
| C. elegans | GR2260 | cdo-1(mg622) X | CGC |
| C. elegans | USD959 | moc-6(rae296) III; cdo-1(mg622) X | This work |
| C. elegans | USD960 | cth-2(mg599) II; moc-6(rae296) III | This work |
| E. coli | BW25113 | Wild type | NIG |
| E. coli | JW0764-2 | ΔmoaA753::kan | NIG |
Acknowledgments
We thank the Caenorhabditis Genetics Center for providing C. elegans strains. We thank the National Institute of Genetics, National BioResource Project (NIG, Japan) for providing E. coli strains.
References
Funding
This work was funded by an NIH grant to Gary Ruvkun (2R01GM044619-29), a Damon Runyon Fellowship to Kurt Warnhoff (DRG-2293-17), as well as an NIH grant to Kurt Warnhoff (5P20GM103620-09 7140).
Reviewed By
Stephan von ReussHistory
Received: November 22, 2021Revision received: February 15, 2022
Accepted: February 17, 2022
Published: February 22, 2022
Copyright
© 2022 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Snoozy, J; Breen, PC; Ruvkun, G; Warnhoff, K (2022). moc-6/MOCS2A is necessary for molybdenum cofactor synthesis in C. elegans. microPublication Biology. 10.17912/micropub.biology.000531.Download: RIS BibTeX
