
Description
Induced protein dimerization is a useful tool to study protein function. A well-established method takes advantage of the binding between the FKBP12 protein (FK506 binding protein 12 kDa) and the FRB domain of the mTOR kinase upon interaction with rapamycin (Putyrski and Schultz 2012). Recently we established a rapamycin-inducible dimerization system for the germ line and early embryos of C. elegans (Mangal et al. 2018). We demonstrated the translocation of mCherry::FKBP12 to the plasma membrane via rapamycin induced binding to FRB::GFP::PH (anchored to the plasma membrane). In order to study the function of a native protein upon rapamycin induced translocation it would be advantageous to tag the genomic region of the gene of interest (GOI) with mCherry::fkbp2 to ensure that the fusion protein is controlled by its native environment.
The CRISPR/Cas9 system has revolutionized genome engineering in C. elegans (Chen et al. 2013; Arribere et al.2014; Paix et al. 2015; Dickinson et al. 2015; Norris et al. 2015; Schwartz and Jorgensen 2016). DNA double-stranded breaks are generated by the endonuclease Cas9, which is guided to its target by a single guide RNA (sgRNA) (Jinek et al. 2012). If a repair donor vector is provided carrying a transgenic sequence flanked by 5’ and 3’ homology regions, the cell can repair these double-stranded breaks via homology directed repair (HDR) by incorporating the transgenic sequence into the cleaved locus. This enables e.g. N- or C-terminal fluorescent protein fusions of the GOI. Drug-selection based screening methods to tag native proteins have been developed (Dickinson et al. 2015; Norris et al. 2015; Schwartz and Jorgensen 2016). One of these streamlined methods uses a dual marker selection cassette (Norris et al. 2015).
Herein, we describe a modification of the dual-marker selection cassette plasmid of Norris et al. (2015) that can be used in conjunction with CRISPR/Cas9, TALEN or Zinc Finger Nucleases to tag endogenous proteins for inducible translocation to the plasma membrane during early embryogenesis or in the germ line.
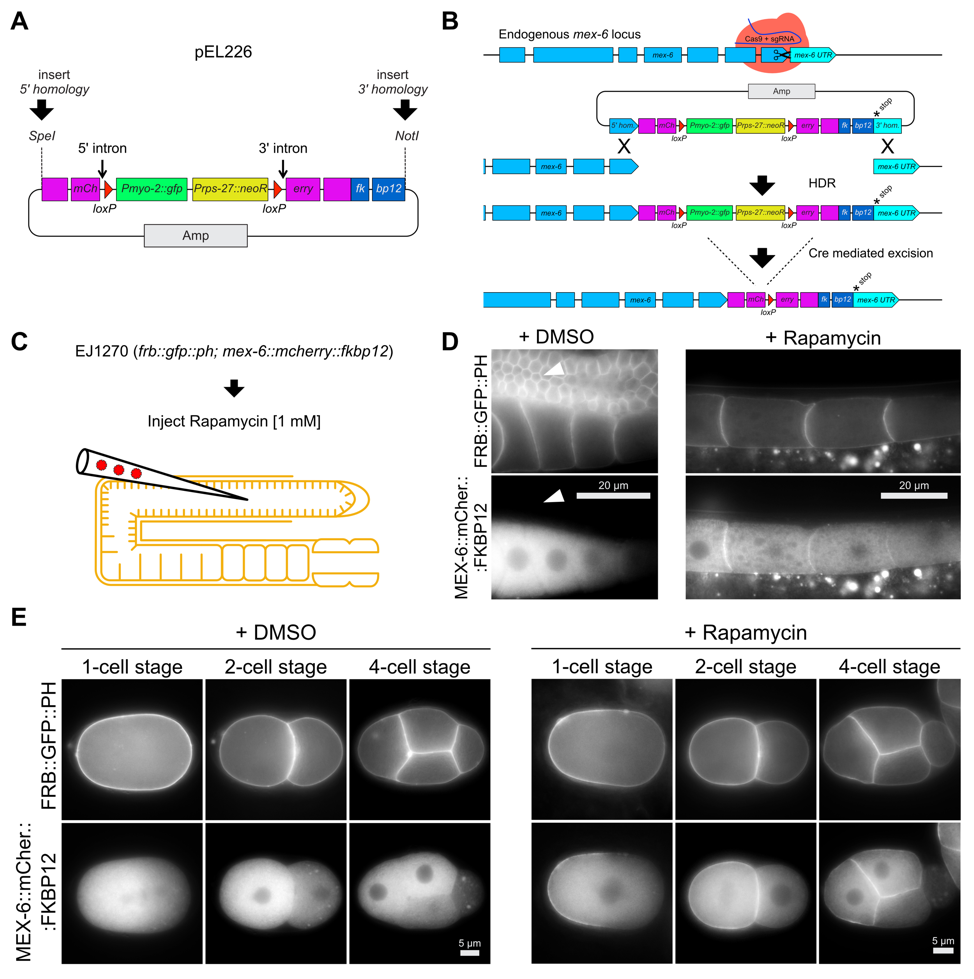
We modified Norris’s mCherry-tag repair donor vector by fusing a C. elegans codon-optimized fkbp12 sequence 3’ to mCherry (Mangal et al. 2018) and re-establishing the critical NotI site for 3’ homology region insertion (Figure 1 A; pEL226). This vector can be used to C-terminally tag any GOI by following the protocol of Norris et al. (2015), including subsequent Cre-mediated excision of the dual marker selection cassette (Figure 1 B). As proof of principle we C-terminally tagged the locus of mex-6 on chromosome II with mCherry::fkbp12 (Figure 1 B). We observed increasing cytoplasmic mCherry signal in late-stage oocytes, and anterior enrichment in early-stage embryos (Figure 1 D and E; DMSO control). We did not detect any mCherry signal in the pachytene region of adult stage gonads (Figure 1 D arrowheads; DMSO control). These observations confirm previous localization studies for mex-6(ax2065[mex-6::gfp]) II transgenic animals (Paix et al. 2014), and mirror those for observed for the paralogous protein, MEX-5 (Schubert et al. 2000; Griffin et al. 2011). We crossed the MEX-6::mCherry::FKBP12 expressing strain (EJ1269, Table 2) with a strain that expresses FRB::GFP::PH (ZAN87; Mangal et al., 2018), which localizes to the plasma membrane in the germ line and early embryos (Figure 1 D and E), and then singled F2 hermaphrodites to obtain a strain that is homozygous for both insertions (EJ1270, Table 2). Animals from this strain were injected with 1 mM rapamycin into the pachytene region of the germ line to induce binding between the FRB and FKBP12 domains. As expected, we observed strong accumulation of MEX-6::mCherry::FKBP12 signal at the plasma membrane of late-stage oocytes and anterior blastomeres of early embryos, 6 h after injection (Figure 1 D and E). Importantly, the MEX-6::mCherry::FKBP12 remains cytoplasmic if DMSO is injected into the germ line as a control (Figure 1 D and E).
Our repair donor vector can be easily modified to tag any GOI with mCherry::fkbp12 in a well-established and streamlined manner (Norris et al. 2015) and it expands the C. elegans CRISPR/Cas9 toolbox for the rapamycin-inducible dimerization system. By crossing into the strain that expresses FRB::GFP::PH (ZAN87) (Mangal et al. 2018) it becomes possible to translocate an endogenously tagged mCherry::FKBP12 protein to the plasma membrane of early embryos or the germ line. Additionally, MEX-6::mCherry::FKBP12 could be used to enrich a protein-of-interest that is tagged with FRB::GFP within the anterior region of early embryos. 2018) it becomes possible to translocate an endogenously tagged mCherry::FKBP12 protein to the plasma membrane of early embryos or the germ line. Additionally, MEX-6::mCherry::FKBP12 could be used to enrich a protein-of-interest that is tagged with FRB::GFP within the anterior region of early embryos.
Reagents
Standard methods for DNA amplification, analysis and manipulation were used. PCR products were amplified by using Phusion® High-Fidelity DNA Polymerase (New England Biolabs), according to the manufacturer’s protocol. DNA sequences were obtained by Sanger sequencing.
We inserted a C. elegans codon optimized sequence of fkbp12 (plus a flexible linker 5’ of fkbp12, gcaggtggaggtact) into the unique NotI site of loxP_myo2_neoR_mCherry_intron (Norris et al. 2015) via Gibson assembly (Gibson et al. 2009) by creating a new unique NotI site 3’ of fkbp12. The resulting vector pEL226 is a universal repair donor vector to tag any locus with mCherry::fkbp12. We digested pEL226 with SpeI and NotI and subsequently column purified the DNA. We amplified ~575 bp 5’ and 3’ homology region of the C-terminus of mex-6 from genomic DNA (3’ homology region includes the stop codon of mex-6 exon7, PAM(mex-6.1) was mutated to NCA) with primers carrying homology tails (Table 1) to the unique SpeI and NotI sites of pEL226. PCR products were subsequently column purified. All four fragments (PCR products plus pEL226 fragments) were fused together via Gibson assembly (pEL228).
We designed sgRNA(mex-6.1) via the online sgRNA design tool http://crispr.cos.uni-heidelberg.de/index.html(Stemmer et al. 2015). sgRNA(mex-6.1) is almost identical to sgRNA18 (Paix et al. 2014) but shifted by 1 nt 3’ and therefore using another PAM. sgRNA(mex-6.1) expression plasmid (pEL227) was cloned by using pRB1017 following the protocol of (Arribere et al. 2014). All plasmids were purified by using PureLink™ HQ Mini Plasmid DNA Purification Kit (Invitrogen) and eluted with Ultrapure Water for Molecular Biology (Merck / EMD MilliporeSigma). We used the following injection-mix concentrations (Norris et al. 2015): Peft-3::Cas9_SV40_NLS::tbb-2_UTR (Peft-3Cas9 expression plasmid) at 50 ng/µl, sgRNA(mex-6.1) plasmid (pEL227, based on pRB1017) at 100 ng/µl, repair donor vector (pEL228) at 50 ng/µl (based on pEL226), pCFJ90 (Frøkjaer-Jensen et al. 2014) at 2.5 ng/µl (Pmyo-2mcherry), pCFJ104 (Frøkjaer-Jensen et al. 2014) at 5ng/µl (Pmyo-3mcherry). Ultrapure Water for Molecular Biology (Merck / EMD MilliporeSigma) was added to a total volume of 10 µl.
We injected 62 young adults (N2) and obtained 7 independent lines. We proceeded with one line and injected a Cre recombinase expression vector (pDD104). We used the following injection-mix concentrations: pDD104 (Dickinson et al. 2013) at 50 ng/µl and pCFJ90 at 2.5 ng/µl. After successful Cre-mediated excision of the dual marker selection cassette (Norris et al. 2015), we obtained the strain EJ1269 (Table 2) showing mCherry signal consistent with GFP signal observed in mex-6(ax2065[mex-6::gfp]) II (Paix et al. 2014). Finally we crossed EJ1269 into ZAN87 (Mangal et al. 2018) resulting in EJ1270 (Table 2).
The rapamycin-induced dimerization was performed according to (Mangal et al. 2018). Microscopy was performed 6 h after injection. Worms were dissected with 20 gauge hypodermic needles in M9 to release embryos. Whole worms were immobilized with 10 mM levamisole. Whole worms or embryos and were mounted on 4% agarose pads. Animals were imaged by using Zeiss Axioskop 2 and MetaMorph software (Molecular Devices). Image processing was performed in Fiji/imageJ 2.0.0 (Schindelin et al. 2012), brightness and contrast were adjusted either in Fiji/imageJ 2.0.0 or Affinity Designer 1.6.1 (Serif).
Table 1: Primer homology tail sequences for cloning homology arms into pEL226
|
Primer homology tails
|
homology tail sequence
|
|
forward primer 5’ homology (SpeI)
|
aaacgacggccagtgaattca
|
|
reverse primer 5’ homology
(SpeI, introducing an alanine 5’ of mCherry) |
cttcttcaccctttgagaccatagc
|
|
forward primer 3’ homology
(NotI, introducing an alanine 3’ of fkbp12, [optional stop codon]) |
cgagctcctcaagctcgaggct[tag]
|
|
reverse primer 3’ homology (NotI)
|
tgattacgccaagcttgcg
|
Table 2: Used C. elegans strain
|
Strain
|
Genotype
|
References
|
|
ZAN87
|
estSi50[pEZ156;pmex-5::frb::gfp::ph::tbb2; Cbr-unc-119(+)]I; unc-119(ed3) III
|
(Mangal et al. 2018)
|
|
EJ1269
|
mex-6(dx203[mex-6::mcherry::fkbp12 + loxP]) II
|
This study
|
|
EJ1270
|
estSi50[pEZ156;pmex-5::frb::gfp::ph::tbb2; Cbr-unc-119(+)]I;
mex-6(dx203[mex-6::mcherry::fkbp12 + loxP]) II[tag] |
This study
|
Acknowledgments
We thank John Calarco for providing repair donor vector loxP_myo2_neoR_mCherry_intron (Norris et al. 2015).
References
Funding
E.J.L. was supported by DFG grant LA3380/2, and this work was also supported by the Emmy-Noether-Program (ZA619/3-1) from the DFG to E.Z.
Reviewed By
Adam NorrisHistory
Received: April 12, 2018Accepted: April 16, 2018
Published: April 17, 2018
Copyright
© 2018 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Zielich, J; Mangal, S; Zanin, E; Lambie, EJ (2018). Establishment of a CRISPR/Cas9-based strategy for inducible protein dimerization. microPublication Biology. 10.17912/W2208R. Erratum in: microPublication Biology. 10.17912/micropub.biology.000195.Download: RIS BibTeX
