Integrated DNA Technologies, Inc., Redwood City, CA 94065, USA

Description
RNA-seq is widely used for the quantitative analysis of transcriptomes in the context of studies of gene expression and regulation (Mortazavi et al., 2008; Ozsolak and Milos, 2011; Wang et al., 2009). Generally, RNA-seq protocols employ poly(A) selection for mRNA enrichment. However, poly(A) based enrichment is subject to potential bias depending on the poly(A) status of various mRNAs, which could be particularly undesirable in the context of studying post-transcriptional gene regulatory mechanisms, such as miRNA repression (Wu et al., 2006). Therefore, ribosomal RNA (rRNA) depletion is a desirable alternative strategy to enrich for mRNA sequences in RNA-seq sample preparation (Zhao et al., 2014). However, currently available rRNA depletion toolkits were designed for either mammals or bacteria, and hence do not offer an efficient option for rRNA depletion of RNA samples from certain experimental organisms, such as C. elegans.
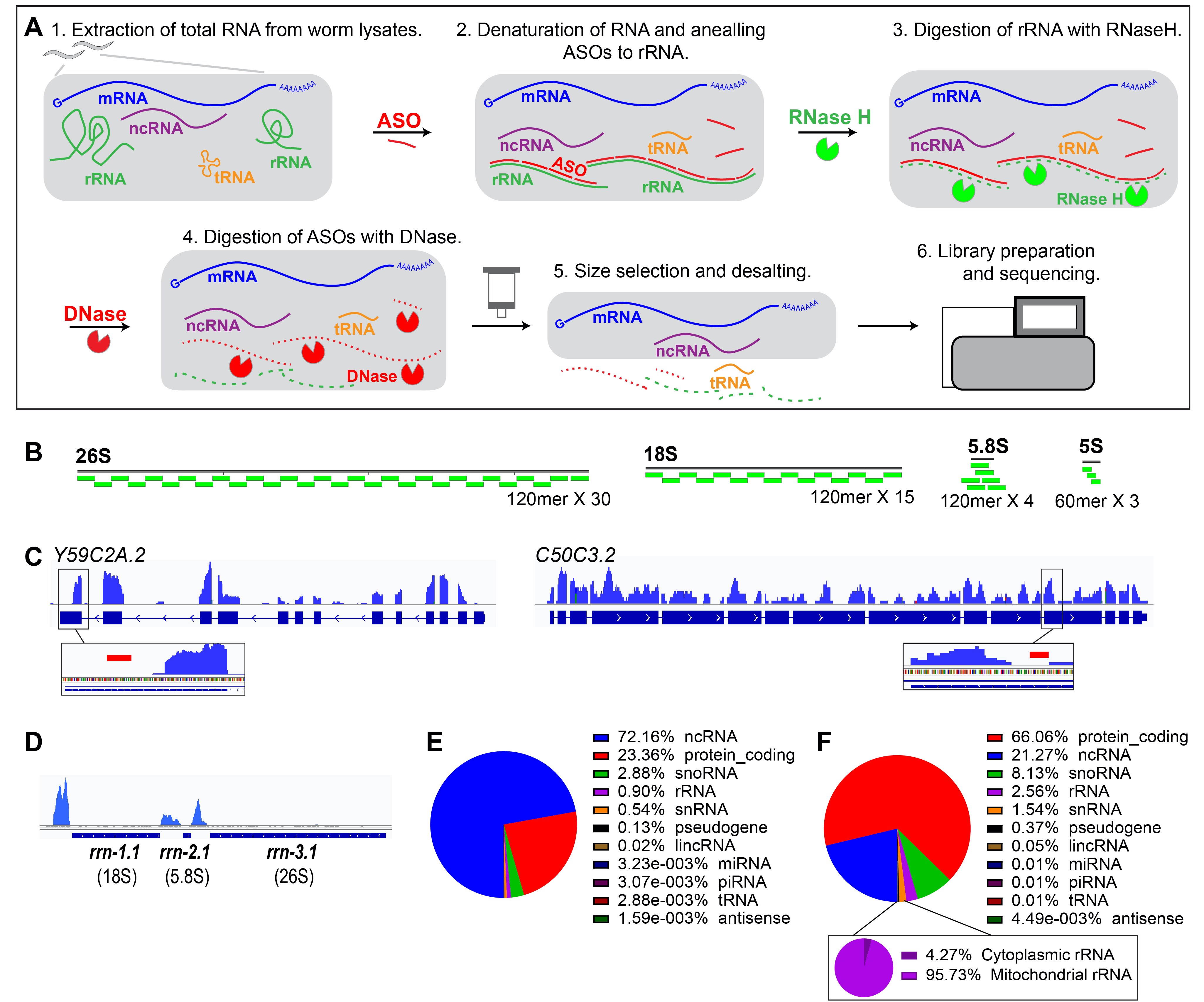
Here we describe a rRNA depletion protocol based on RNase H digestion using antisense oligonucleotides (ASOs) specifically designed for C. elegans cytoplasmic rRNA (Fig. 1A). We suggest that this rRNA depletion protocol is applicable to RNA-seq applications where the yield of mRNA enrichment should be independent of poly(A) status, or any application which benefits from the removal of rRNA sequences, such ribosome profiling, or sequencing of non-coding RNAs other than rRNA.
We designed 120mer DNA ASOs complementary to 26S, 18S and 5.8S rRNA sequences and 60mer ASOs complementary to 5S rRNA (Fig. 1B). We used thermostable RNase H and optimized protocols at high temperature for the ASO-rRNA duplex annealing and digestion to reduce the formation of secondary structure of rRNA and off-target annealing of the ASOs. We found that among the 12 RNA-seq libraries (4 different C. elegans genotypes) prepared with our rRNA depletion protocol, the cytoplasmic rRNA (26S, 18S, 5.8S and 5S) reads only comprised < 0.2 % of the total reads (Fig. 1E-F), indicating that our protocol is effective for removing rRNA. Highlighting the specificity of the depletion, we observed reads from rRNA precursor transcripts (Fig. 1D) and mitochondrial rRNA (Fig. 1F, lower panel) due to the absence of ASOs against those sequences. As expected, the rRNA-depleted datasets contained a large representation of other non-coding RNA sequences, further supporting that our protocol enriches for RNAs lacking poly(A) (Fig. 1E-F). The performance of our depletion protocol appears to be similar to that of two independently developed similar protocols for C. elegans-specific rRNA depletion (Arribere et al., 2016; Barucci et al., 2020). Interestingly, we found that a large proportion of the non-coding RNA reads after RNA depletion correspond to the signal recognition particle RNA (srpR) (Regalia et al., 2002). A similar (although somewhat less prominent) enrichment of srpR was also observed in the libraries of Barucci et al., 2020 and Arribere et al., 2016. The srpR reads were manually removed in our analysis (Fig. 1F); however we suggest that later iterations of our protocol could include ASOs corresponding to srpR, as well as mitochondrial rRNA and cellular primary rRNA transcripts.
Although we did not directly compare the genome-wide distribution of mRNA reads from this ASO protocol to that of mRNA reads from other approaches for mRNA enrichment, we did conduct a computational assessment of the potential for off-target effects of our ASO rRNA depletion. We adopted a conservative assumption that, for an ASO to trigger RNase H digestion of an mRNA sequence, it should match the mRNA with a melting temperature of no less than 20oC below the temperature at which the annealing and RNase H digestion were performed. Using BLAST, we identified only two potential matches between the ASOs and the C. elegans transcriptome that met this criterion (Morgulis et al., 2008). Interestingly, these two sites do seem to locate in regions of their respective transcriptional units where reads were relatively depleted locally, suggesting that the ASOs may have triggered off-target depletion at these sites (Fig. 1C). However, the apparent sequence depletion in these two instances was only restricted to regions around the ASO off-target match sites, and the alignment distribution for each gene in sequences other than the off-target sites seems to be relatively unaffected (Fig. 1C). From this analysis, we suggest that the ASO rRNA depletion method results in essentially negligible off-target mRNA depletion.
Methods
Request a detailed protocolrRNA depletion and RNA-seq
Harvested worms were washed with M9 medium and flash-frozen in liquid nitrogen. The worm pellets were lysed by QIAzol (Qiagen, Cat: 79306) as previously described (McJunkin and Ambros, 2017). ASOs and total RNA were mixed with approximate 2:1 molar ratio (the quantity of ASOs calculated assuming rRNAs comprise > 90% of total RNA). Higher molar ratios of up to 6.5:1 were tested but did not result in observable improvement in depletion efficacy. Accordingly, a mixture containing final concentrations of 0.1 mM of each ASO, 50 ng/µl total RNA, 10 mM Tris-HCl (pH = 8.0), 100 mM NaCl, 1 mM EDTA and 0.8 U/µl RNase Inhibitor (NEB, Cat:M0314) was incubated at 95 °C for 2 min for denaturation and then 65 °C for 30 min for annealing. We have also tested annealing conditions with gradual decrease in temperature (0.5-2 °C per minute) but no apparent improvement in depletion efficacy was observed. To digest the rRNA, thermostable RNase H and the buffer (MCLAB, Cat:HTRH-100) were preheated to 65 °C and added to the reaction (while maintaining the reaction at 65 °C) to obtain a final concentration of 0.2 U/µl RNase H and the reaction was further incubated at 65 °C for 40 min, and then chilled on ice. Other digestion temperatures from 30 to 85 °C were also tested, and 65 °C was determined to be optimal for reducing rRNA secondary structure without compromising overall RNA integrity of the samples. To digest the ASOs, Turbo DNase (Invitrogen, Cat:AM2238) and the buffer were added to a final concentration of 0.1 U/µl and the reaction was incubated at 37 °C for 25 min. mRNA was then purified by RNA Clean & Concentrator-5 Kit (ZYMO, Cat:R1015) as described in (Zhang et al., 2012). The RNA-seq libraries were constructed by NEBNext Ultra II RNA Library Prep kit (NEB, Cat:E7775, E7335, E7500) and sequenced by Illumina NextSeq 500 system.
Data analysis
Adaptor sequences were trimmed and reads shorter than 15 nt were filtered out from analysis by Cutadapt/1.4.1 (Martin, 2011). For the analysis shown in Fig. 1F, the srpR reads were removed by initially mapping with Bowtie2/2.3.4.3 (Langmead and Salzberg, 2012), and either total filtered reads (Fig. 1E) or the remaining reads after srpR removal (Fig. 1F) were mapped to C. elegans genome (WBcel235) by Star/2.5.3 with default parameters (Dobin et al., 2013). Mapping data was sorted and processed by Samtools/1.9 (Li et al., 2009). Gene counting was performed using featureCounts (Liao et al., 2014).
Reagents
The ASO sequences in this research are available upon request.
Acknowledgments
We thank Caifu Chen and Nicole Sponer at Integrated DNA Technologies, Inc. for help in developing the rRNA depletion protocol. We thank Isana Veksler-Lublinsky from Ben-Gurion University of the Negev, Beer-Sheva, Israel for advice on data analysis.
References
Funding
NIH grants R01GM088365, R01GM034028, and R35GM131741 (VA)
Reviewed By
AnonymousHistory
Received: September 11, 2020Revision received: September 20, 2020
Accepted: September 21, 2020
Published: September 22, 2020
Copyright
© 2020 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Duan, Y; Sun, Y; Ambros, V (2020). RNA-seq with RNase H-based ribosomal RNA depletion specifically designed for C. elegans. microPublication Biology. 10.17912/micropub.biology.000312.Download: RIS BibTeX
