
Abstract
Saul-Wilson Syndrome is an ultra-rare skeletal syndrome caused by a mutation in the COG4 gene resulting in a glycine-to-arginine substitution at amino acid position 516. The COG4 gene encodes one of 8 subunits of the conserved oligomeric Golgi complex. Using CRISPR-Cas9, our lab generated a C. elegans model for Saul-Wilson Syndrome by recreating the same glycine-to-arginine substitution in the worm ortholog cogc-4. Upon observation, the cogc-4(av107) worms did not display any obvious differences compared to wild-type worms. We used a variety of assays including stressing the worms using heat and Paraquat, as well as RNAi against the 7 other COG complex subunit genes in an attempt to uncover a phenotype. Our data suggest that this mutation in cogc-4(av107) worms does not lead to a detectable phenotype. Further studies should aim at more directly assessing Golgi function in this disease model.
Description
Saul-Wilson Syndrome (SWS) is an ultra-rare, autosomal dominant skeletal dysplasia syndrome discovered in 1990; only 16 patients have been identified to date (Saul and Wilson 1990; Ferreira et al. 2018, OMIM#: 618150). The disease is characterized by short stature, various craniofacial abnormalities, shortened fingers and toes, and speech and physical developmental delay (Ferreira 2020). SWS is caused by a missense mutation in the COG4 gene, resulting in a G516R residue change. Other pathogenic mutations have been observed in this gene and all are clustered at the C-terminal end of the protein (R724W, R729W, R729A, E764A). These are associated with Congenital Disorder of Glycosylation type 2j (CDGIIj). This is a recessive disease characterized by mild psychomotor delay, mild dysmorphic features, epilepsy, and defective sialylation (Reynders et al. 2009). Besides the mild developmental delay, this disease seems to share virtually no phenotypic similarity with SWS.
Human COG4 is one of 8 protein subunits that comprise a bi-lobed complex known as the Conserved Oligomeric Golgi Complex or COG complex (Smith and Lupashin 2008). This complex is involved in intra-Golgi trafficking and glycoprotein modification (Reynders et al. 2009). All COG genes, with the exception of COG3, have been implicated in recessive Congenital Disorders of Glycosylation (CDGs).
In assessing the clinical phenotype, Ferreira and others (2018) have reported data from a cohort of 14 SWS patients. These individuals did not have obvious glycosylation defects in serum proteins analyzed, although the secreted proteoglycan decorin appeared to be at a higher molecular weight than decorin secreted from control cells. This suggests some types of Golgi processing compartments are affected in SWS. Besides this observation, cellular trafficking in fibroblast cultures appeared normal and there was also no defect in COG subunit mRNA, protein expression, or localization. Immunofluorescence showed that nearly half of SWS patient fibroblasts had abnormal Golgi morphology characterized by collapsed stacks, while control fibroblasts had over 93% normal Golgi stacks. After treatment with Brefeldin A (BFA), a compound known to inhibit transport between the Golgi and ER, patient cells appeared to have a collapsed Golgi nearly twice as soon after treatment compared to control cells. This was evidenced by cis- and trans-Golgi markers becoming co-localized, and was termed “accelerated anterograde transport”– thus, attributing gain-of-function status to this G516R mutation in the mutant COG4 protein (Ferreira et al. 2018).
Two zebrafish models homozygous for frameshift mutations and both resulting in a truncated COG protein also revealed morphological Golgi defects, as well as inner ear deformities and decreased number of hair bundles in mechanosensory hair cells (Ferreira et al. 2018). In addition, biochemical findings included defects in proteoglycan secretion, as well as some types of collagen having decreased expression in certain tissues. These findings in the zebrafish models as well as from patient cultures have established a role for COG4 in inner ear development and skeletogenesis, which is, through some unknown mechanism, associated with defective Golgi structure. To this date, there has not been a model organism model studied in which the conserved Saul-Wilson allele has been mutated.
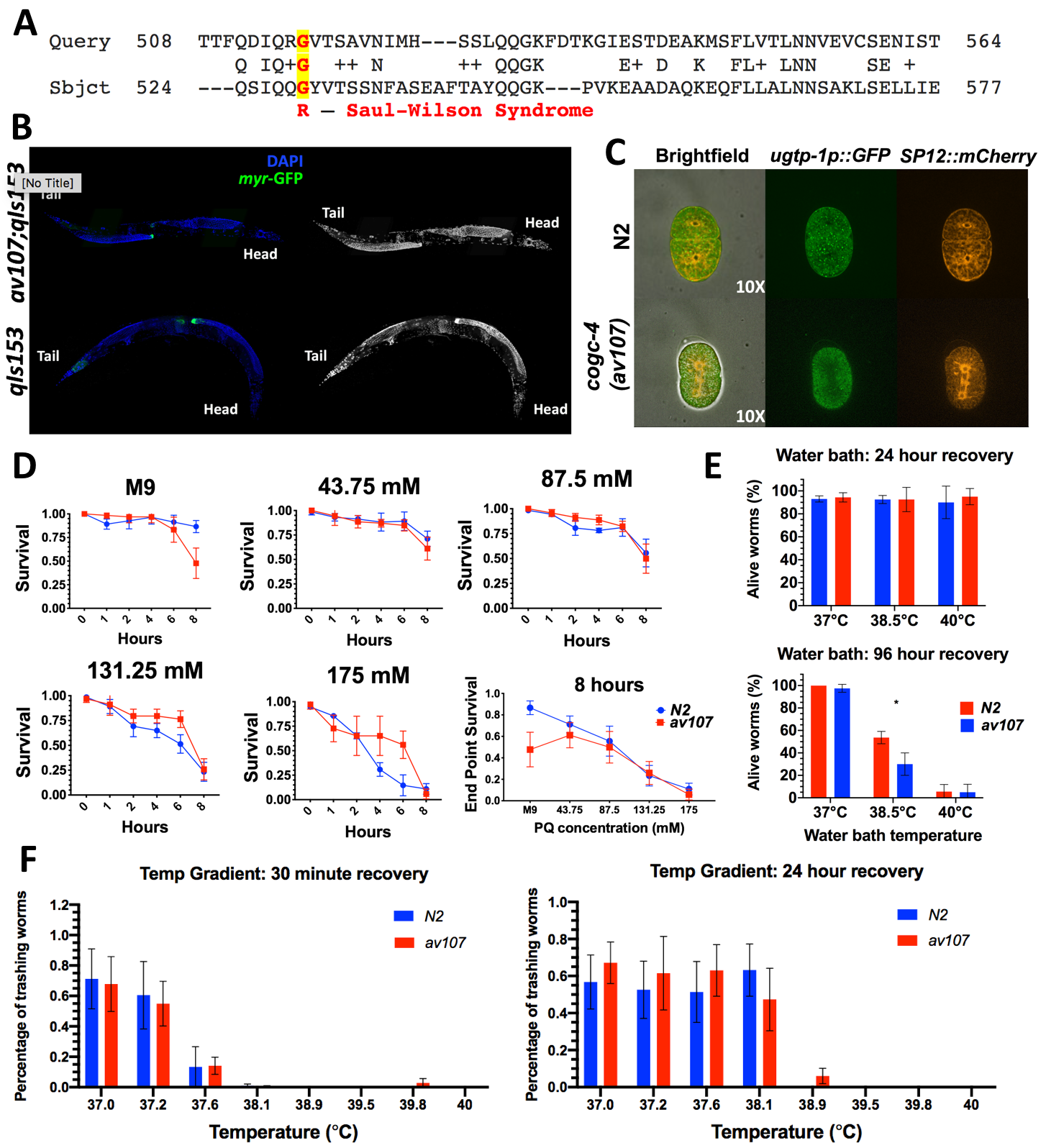
Given the moderate degree of amino acid conservation (29% amino identity, UniProt; 65.7% similarity, LALIGN) between human COG4 and the C. elegans ortholog COGC-4 (Fig. 1A), we decided to recreate the Saul-Wilson allele in the worm and assay for any Golgi-related phenotypes. On a macroscopic level, the worms did not display any morphological, reproductive, locomotive, or other behavioral defects. In addition, RNAi of each of the other 7 COG subunit genes individually did not reveal an obvious phenotype in these cogc-4(av107) worms as they were phenotypically indistinguishable from N2 worms. However, it should be noted that gene knockdown quantification was not verified with qRT-PCR, therefore there is the potential for incomplete knockdown.
It has been reported that RNAi of the individual COG complex lobe 1 subunit genes (cogc-1, cogc-2, cogc-3 and cogc-4, respectively) reveal gonad arm and distal tip cell (DTC) migration defects (Kubota et al. 2006). Therefore, we crossed in a DTC marker, but the av107 worms did not display distal tip cell migration defects using the distal tip cell marker, despite the cogc-4 RNAi DTC migration defect phenotype reported in Kubota et al. 2006 (Figure 1B). We also crossed in fluorescent Golgi and ER markers to monitor for evidence of Golgi collapse. Normal ER and Golgi morphology and no evidence of co-localization was observed in our cogc-4(av107) early embryos (Fig. 1C).
Given its role in Golgi trafficking, we then decided to perform a variety of stress assays—using heat and oxidative stress— in an attempt to uncover a phenotype (Fig. 1D-F). We reasoned that stressing worms could exacerbate a stronger and quantifiable phenotype. We used the methods described in Senchuk et al. (2017) as a reference in addition to troubleshooting to optimize the conditions such as Paraquat concentration and exposure time as well as optimal temperature ranges that worked best for our assays. There did not appear to be a difference in both survival after heat shock or recovery after heat shock for embryos or L4 stage worms between the two genotypes at any of the conditions tested. This is also true for survival in the presence of Paraquat (PQ) (Fig. 1D). It should be noted that all assays were performed using animals homozygous for the G529R mutation. We will continue to test these animals for phenotypes under a variety of other stresses and challenges.
Methods
Request a detailed protocolGeneration of Saul-Wilson Allele using CRISPR: A single guide RNA (sgRNA) was designed that targeted the region surrounding position G529 in cogc-4 (5’–ATGAGGTGACGTATCCCTGC– 3’). The DNA repair oligo (5’–GATACAGTCGTCCACGGGCATTTACGCCAGAGCATACAACAACGTTACGTCACCTCATCCAATTTCGCATCTGAAGCATT– 3’) was designed to contain a mutated PAM site, 3 silent mutations, the desired G529R change and a restriction site for AclI used in genotyping for edits (underlined in the sequence shown). Missense mutations were identified using the following primers: forward 5‘- CTCAATCGTCGACGATGTGG; and reverse 5’- TTGATAGTGTCTGTGCCACC.
Heat Shock: Water Bath: A preliminary assay was performed using bleach-synchronized embryos being treated at 37°C or 38.5°C for 30 minutes or 180 minutes. Embryos were checked for viability and development into fertile adults 24 and 96 hours following the assay. However, at 37°C, 100% of N2 and cogc-4(av107) embryos hatched and developed into fertile adults after both the 30 minute and 180 minute exposure times. At 38.5°C, 100% of N2 and cogc-4(av107) embryos died after both 30 minute and 180 minute exposure times. We then decided to assay L4 stage worms in order to perhaps see more subtle phenotypes. Bleach-synchronized worms were allowed to reach L4 stage and were then incubated for 3 hours at 37°C, 38°C, or 40°C in a water bath. L4s were counted immediately after heat shock, and allowed 24 hours for recovery at 20°C, as well as assessed 96 hours after the heat shock treatment. Worms were assessed by eye for viability and movement by either poking them with a pick or by observing thrashing after a 1 ul drop of M9 was pipetted onto them on the agar plate.
Heat Shock: Temperature Gradient: Approximately ~1000 embryos were synchronized and placed onto multiple plates. Once the worms reached the L4 stage, worms were rinsed off of the plate(s) in a total of 2 ml of M9 and were added to two (2) separate Eppendorf tubes. 30 ul of worms in M9 were added into each of the tubes of an 8-tube PCR strip. Tubes were placed in the thermocycler and a heat gradient was run for 30 minutes that was programmed to the following eight temperatures: 37.0°C, 37.2°C, 37.6°C, 38.1°C, 38.9°C, 39.5°C, 39.8°C, and 40°C. Five replicates were performed for both genotypes (N2 and av107). After the 30 minutes, the contents of each tube were pipetted and aliquoted into a 96-well plate for assessment of survival. Worms were counted and assessed for thrashing or signs of viability 30 minutes after heat shock and 24 hours after the shock. They were gently poked with a platinum wire tip to check for movement.
Paraquat Survival Assay: cogc-4(av107) and N2 populations were bleach-synchronized, plated, and the embryos allowed to develop until the desired stage had been reached (L3-young L4s). Paraquat (PQ) solutions were prepared from a 200 mM stock, diluted with M9 to get final concentrations of 43.75 mM, 87.5 mM, 131.25 mM and 175 mM PQ solutions (accounting for the 5 ul of worms in M9 added to each well in later steps). 35 ul of each solution was added to the corresponding wells of a 96-well plate. Worms were washed off the plates using M9 and into an Eppendorf tube. M9 was either removed or added in order to make the final concentration of worms ~1 worm/ul. Worms were observed immediately after being added to the paraquat solution (time point 0), after 1 hour (T1), 2 hours (T2), and then every 2 hours until 8 hours had passed since being placed in the wells (T3-T5). At each time point, each well was stirred for about 3 seconds with a wooden toothpick before the number of moving worms per well was counted. Worms were counted as “alive” if visibly thrashing, or if any type of head movement was observed following “stirring” or directly after being gently poked with the wooden stick. Similarly, worms were counted as “dead” if no movement at all was observed after being stirred or poked directly. Total worms per well were counted at the beginning of the assay (immediately following the T0 viability count) and after the T5 counts.
Statistical analyses were performed for all Paraquat assay plots and temperature assays using GraphPad Prism 9.0 Software.
Microscopy: Images were captured by high-resolution confocal microscopy with Nikon 60 X 1.2 NA water objective with 1μm z-step size, and 60% intensity for the 488 nm and 561 nm lasers. Images were generated by custom Fiji Image J>Stacks>Z project (Linkert et al. 2010, Schindelin et al. 2012).
Reagents
Strains: The Saul-Wilson allele strain we generated by CRISPR is AG529: cogc-4(av107) G529R. The DTC marker was crossed in from strain JK4475 (qIs153 [lag-2p::MYR::GFP + ttx-3p::DsRed] V). The ER marker used was crossed in from strain OCF15: pie-1p::mCherry::sp12::pie-1 3’UTR + unc-119(+). The Golgi marker used was crossed in from strain WH351: pie-1::GFP::ugtp-1 + unc-119(+). The three marker strains were obtained from the CGC.
Paraquat Assays: Paraquat dichloride hydrate was purchased from Millipore Sigma (36541-100MG).
Acknowledgments
We thank the Caenorhabditis Genetics Center for providing strains and Dr. Carlos Ferreira for encouraging us to generate this worm model for SWS.
References
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. Some C. elegans strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Reviewed By
Jennifer KowalskiHistory
Received: November 16, 2020Revision received: February 21, 2021
Accepted: February 22, 2021
Published: March 4, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Zafra, I; Nebenfuehr, B; Golden, A (2021). Saul-Wilson Syndrome Missense Allele Does Not Show Obvious Golgi Defects in a C. elegans Model. microPublication Biology. 10.17912/micropub.biology.000373. Corrigendum in: microPublication Biology. 10.17912/micropub.biology.000379.Download: RIS BibTeX
