
Abstract
Variants of the CACNA1C voltage-gated calcium channel gene have been associated with autism and other neurodevelopmental disorders including bipolar disorder, schizophrenia, and ADHD. The Timothy syndrome mutation is a rare de novo gain-of-function variant in CACNA1C that causes autism with high penetrance, providing a powerful avenue into investigating the role of CACNA1C variants in neurodevelopmental disorders. In our previous work, we demonstrated that an egl-19(gof) mutation, which is equivalent to the Timothy syndrome mutation in CACNA1C, can disrupt termination of the PLM axon in C. elegans. Here, we report a novel phenotype for the egl-19(gof) mutation, whereby it causes the growth of an ectopic process from the ALM cell body. We also extend our previous results to show that the egl-19(gof) mutation causes axon termination defects not only in the PLM axon, but also in the ALM axon. These results suggest that the Timothy syndrome mutation can disrupt multiple steps of axon development. Further work exploring the molecular mechanisms that underlie these perturbations in neuronal polarity and axon termination will give us better understanding of how variants in CACNA1C contribute to the axonal defects that underlie autism.
Description
The egl-19 gene in C. elegans encodes the pore forming subunit for the L-type voltage gated calcium channel that is homologous to the CACNA1C gene in humans (Lee et al., 1997). Variants in CACNA1C are risk factors for autism and other neurodevelopmental disorders (Li et al., 2015; Lu et al., 2012; Strom, et al., 2010). Timothy syndrome is a syndromic form of autism that can be caused by either of three rare de novo mutations in CACNA1C. These mutations cause either a G402R, G402S or G406R mutation in the CACNA1C protein (Splawski et al., 2004; Bader et al., 2011). Our previous work demonstrated that PLM axon termination is disrupted by mutations equivalent to the G402R and G406R mutations in CACNA1C (Buddell et al., 2019). Our study also revealed behavioral defects in these mutant worms. Although the anatomical basis for these behavioral defects has not been determined, it is likely that they are caused by multiple defects within the mechanosensory system.
To learn more about how the Timothy syndrome mutations can alter neuronal development, we focused on the egl-19(n2368) and egl-19(tr134) mutations (Lee et al., 1997; Kwok et al., 2008), hereafter called the egl-19(gof) mutations. Both of these egl-19(gof) mutations lead to a G365R amino acid change in EGL-19 that is equivalent to the G402R gain of function mutation in CACNA1C that can cause Timothy syndrome in humans. Here, we report that the egl-19(gof) mutations can cause the growth of an ectopic process from the cell body of the ALM neuron. This observation suggests that in addition to causing defects in axon termination, the egl-19(gof) mutations can also disrupt the polarity of process outgrowth.
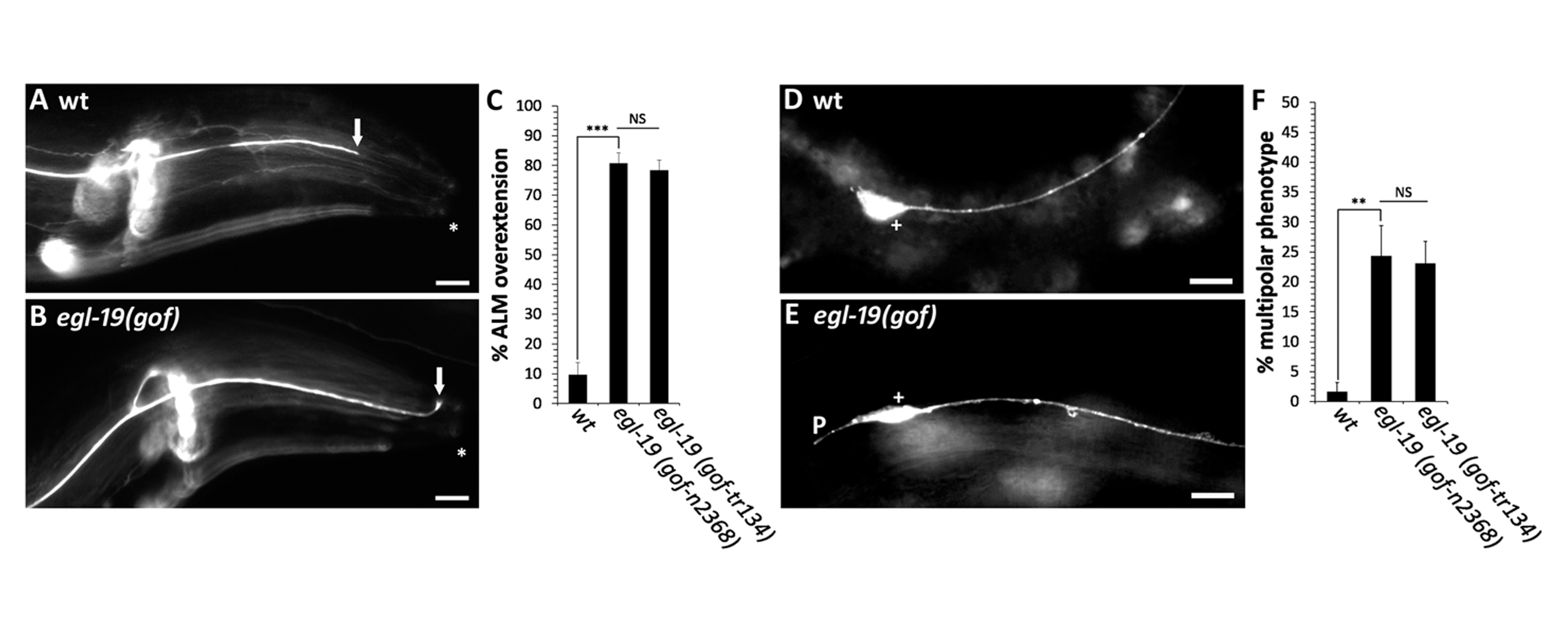
The mechanosensory neurons in C. elegans are responsible for transducing light touch and consist of two ALM neurons, two PLM neurons, one AVM neuron and one PVM neuron (Chalfie et al., 1985). To identify neuronal defects caused by the egl-19(gof) mutations, we labeled each of the six mechanosensory neurons with a fluorescent transgene that is expressed in each of the six mechanosensory neurons. After observing each of the six mechanosensory neurons in populations of egl-19(gof) mutants, we identified a novel phenotype in the ALM neuron. In wild-type animals, nearly all ALM neurons extend a single process from the anterior side of the cell body (Figure 1D,F). However, in egl-19(gof) animals, we often observed a second short process that extended in the posterior direction (Figure 1E,F). In addition to this novel phenotype, we also found an axon termination defect in ALM neurons that is similar to the axon termination defect that we previously reported in the PLM neuron. In wild-type animals, the cell bodies of the ALM neurons reside on the lateral body wall and extend a single axon into the head, where they terminate prior to reaching the tip of the nose (Figure 1A). In egl-19(gof) mutants, we observed overextended ALM axons, where the axon extended past its normal termination point and terminated within the tip of the nose (Figure 1B,C).
These results suggest that the Timothy syndrome mutation can disrupt multiple steps of axon development. First, the egl-19(gof) mutations can disrupt the polarization of process outgrowth. Second, the egl-19(gof) mutations can also disrupt axon termination. Future work will address the molecular mechanisms that underlie these alterations in neuronal polarity and axon termination. An understanding of these mechanisms will be critical to our understanding of how variants in CACNA1C give rise to the axonal defects that underlie autism.
Methods
Request a detailed protocolC. elegans strains were cultured and maintained on nematode growth medium (NGM)-agar plates using standard methods at 20°C (Brenner, 1974). Axons were labeled and observed as previously described (Xu et al., 2012). Briefly, animals were mounted on a 5% agarose pad and observed with a 40x objective. PLM & ALM neurons were visualized with the muIs32 transgene which encodes Pmec-7::gfp + lin-15(+) and is expressed in all mechanosensory neurons (Ch’ng et al., 2003). The microscope used for imaging and phenotype analysis was the Zeiss Axio Imager M2. Images were acquired using an AxioCam MRm camera and were analyzed using Axiovision 4 software.
Reagents
AGC48: muIs32 [mec-7p::GFP + lin-15(+)] II; egl-19(n2368) IV
AGC139: muIs32 [mec-7p::GFP + lin-15(+)] II; egl-19(tr134) IV
Acknowledgments
We would like to thank Peter Roy and the Caenorhabditis Genetics Center for strains.
References
Funding
This work was funded by the National Institute of Mental Health grant R01MH119157 (to CCQ) and by the National Institute of Neurological Disorders and Stroke grant R03NS101524 (to CCQ). This article does not represent the official views of the National Institutes of Health and the authors bear sole responsibility for its content. Additional funding came from a Research Growth Initiative grant #101X356 from the University of Wisconsin-Milwaukee to CCQ, and a Shaw Scientist Award from the Greater Milwaukee Foundation to CCQ. The Caenorhabditis Genetics Center was funded by NIH P40 OD010440. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Reviewed By
Erik LundquistHistory
Received: December 2, 2020Revision received: March 22, 2021
Accepted: March 23, 2021
Published: April 1, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Buddell, T; Quinn, CC (2021). An autism-associated calcium channel variant causes defects in neuronal polarity in the ALM neuron of C. elegans. microPublication Biology. 10.17912/micropub.biology.000378.Download: RIS BibTeX
