Department of Molecular Biosciences, The University of Texas at Austin, Austin TX, USA

Abstract
Plasmid-based CRISPR knock-in is a streamlined, scalable, and versatile approach for generating fluorescent protein tags in C. elegans (Dickinson et al. 2015; Schwartz and Jorgensen 2016). However, compared to more recent protocols that utilize commercially available Cas9/RNP products and linear DNA repair templates (Dokshin et al. 2018; Ghanta and Mello 2020), the cloning required for plasmid-based protocols has been cited as a drawback of this knock-in approach. Using thorough quantitative assessment, we have found that cloning efficiency can reproducibly reach 90% for the plasmids of the self-excising cassette (SEC) selection method, essentially resolving cloning as a burden for plasmid-based CRISPR knock-in.
Description
The Glow Worms research stream is a course-based undergraduate research education (CURE) laboratory that focuses on making fluorescent protein knock-in strains at genetic loci of interest to the research community. To facilitate making strains, we aimed to determine whether the self-excising cassette (SEC) selection method for CRISPR knock-in could be improved. Indeed, we recently reported that using high-quality DNA and reducing Cas9/sgRNA concentration facilitates recovery of homology directed repair (HDR) knock-ins, requiring microinjection of only 10 P0 worms (Huang et al. 2021). Here, we show that cloning of SEC-based CRISPR plasmids can also be highly efficient and reproducible when optimized based on quantitative considerations.
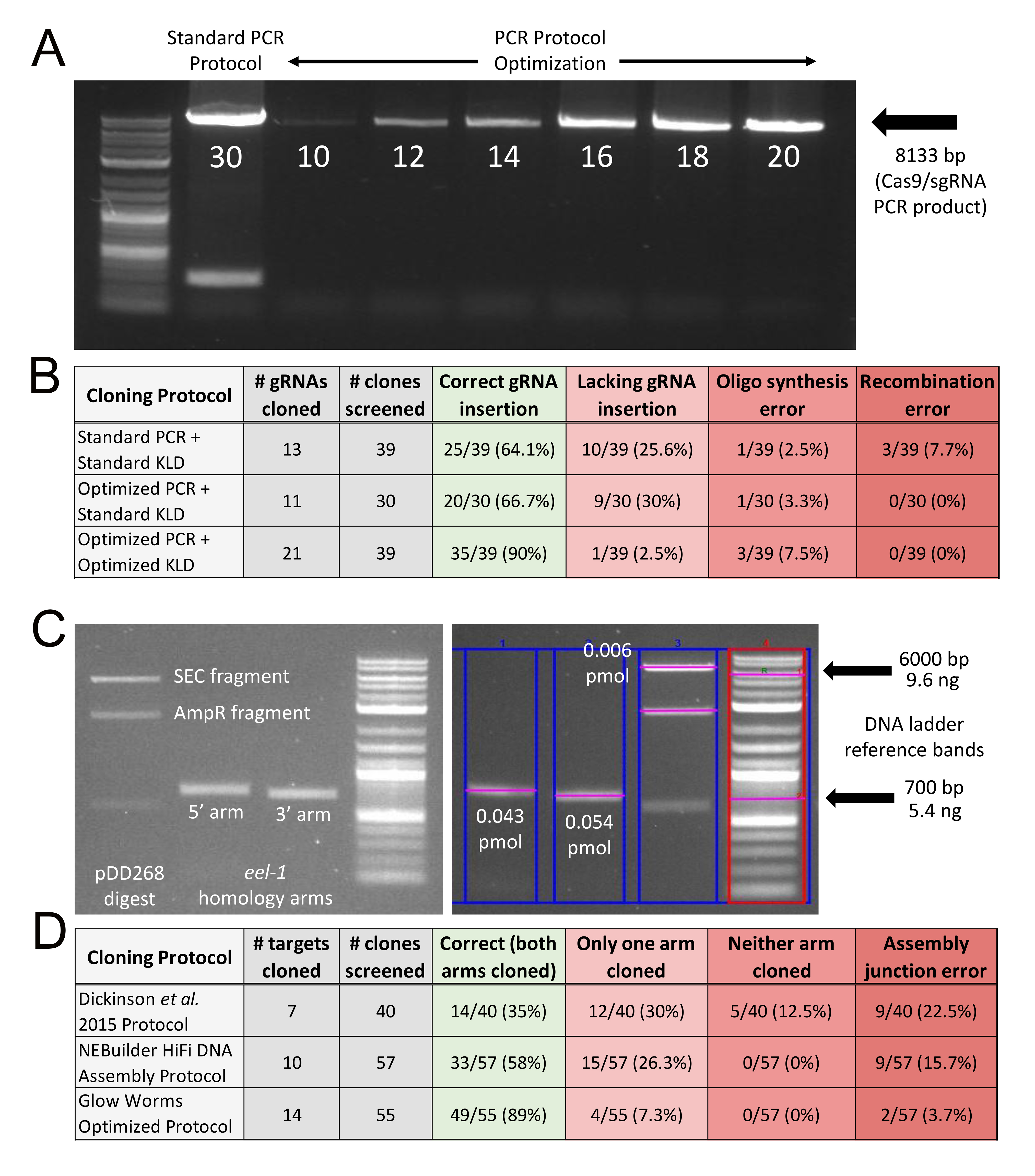
Construction of the Cas9/sgRNA plasmid requires inserting a unique gRNA sequence into plasmid pDD162 (Dickinson et al. 2013) or one of its derivatives (Ward 2015; Aljohani et al. 2020) via site-directed mutagenesis. This involves a PCR step, followed by a KLD ligation step. Using a standard NEB® Q5 PCR protocol of 30 cycles (M0491) and a standard NEB® KLD reaction protocol (M0554), we found that 64.1% of clones included a correct gRNA insertion (Fig. 1A, B). Incorrect clones could be categorized into one of three groups: (1) those lacking the gRNA insertion (i.e. the pDD162 PCR template was transformed instead); (2) those lacking a base or two at the 5’ end of the gRNA (i.e. presumably the result of oligos in the primer pool with incomplete synthesis errors); or (3) those where a reproducible recombination of pDD162 had occurred (Fig. 1B). Based on the types of incorrect clones observed, we hypothesized that we were both using too much DNA in the KLD reaction and that the 5 minute room temperature incubation was insufficient for DpnI to digest the methylated pDD162 template prior to transformation. Indeed, limiting the PCR to 15 cycles and inclusion of a 20 minute 37°C incubation step in the KLD ligation protocol resulted in 90% of clones being correct Cas9/sgRNA plasmids (Fig. 1A, B). This is highly cost effective, as we now routinely transform with as little as 10 µL of competent cells and only need to screen one clone.
Construction of the FP/SEC plasmid requires insertion of two PCR-amplified homology arms into a pre-digested plasmid via Gibson assembly (Dickinson et al. 2015). Using the published protocol, which specifies a 4:1 volume ratio of purified homology arms to purified vector DNA (Dickinson et al. 2015), we found that only 35% of clones were correct assemblies, and the percentage of correct clones was highly variable among unique assembly reactions (Fig. 1D). Indeed, sometimes we would need to screen as many as 10 clones to identify a correctly assembled plasmid. This low efficiency was most likely due to both student inexperience with cloning and inconsistencies in the amount of DNA being used in the assemblies. Thus, we decided to take a more quantitative approach, following the recommendations of the NEBuilder® HiFi DNA Assembly protocol for a four fragment assembly, which specifies a 1:1 ratio of homology arms and vector DNA at 0.05 pmol each for all fragments. We quantified the DNA using ethidium bromide staining, an NEB® DNA ladder standard (N3200S), and Bio-Rad Image Lab 6.0 software (Fig. 1C). This definitely resulted in improved efficiency, yielding 58% of clones with correct assemblies (Fig. 1D). Nevertheless, the frequency of assembly errors remained high (42%), so we wondered whether we were again using too much DNA. We reduced the total amount of DNA 16-fold and modified the protocol to a 2:1 ratio of homology arms to vector DNA, which we based on our typical yield of homology arm and vector DNA following spin column clean-up, as well as limiting the total reaction volume to 10 µL (see Methods). Remarkably, this resulted in a highly reproducible success rate, with an average of 89% of clones being correct for 14 unique assemblies representing 14 different gene targets (Fig 1D). Thus, we now routinely need to screen just one or two clones per assembly, and oftentimes 100% of the clones are correct when screening three or more.
Overall, in combination with our previous report of highly improved knock-in efficiency via the SEC selection method (Huang et al. 2021), our new finding that SEC-based plasmids can be reproducibly cloned at 90% efficiency has essentially resolved the two major drawbacks of plasmid-based CRISPR knock-in. As such, plasmid-based approaches are equally attractive relative to recent approaches utilizing commercially available Cas9/RNP products and linear DNA repair templates that do not require any cloning (Dokshin et al. 2018; Ghanta and Mello 2020). As an undergraduate CURE, plasmid-based approaches are more cost effective, much more scalable to a large number of students, an excellent educational opportunity for molecular cloning, and plasmids are a permanent archive of sequence-verified clones that can be relatively easily repurposed for alternative tags such as other fluorescent markers or auxin-inducible degron (AID) (Ashley et al. 2021). We anticipate that our improvements will make plasmid-based CRISPR knock-in accessible for a wider range of researchers.
Methods
Request a detailed protocolList of genes selected for cloning of gRNAs and homology arms in this study (45)
· amph-1, asp-3, asp-4, attf-2, ccdc-47, cdc-37, cey-1, cey-4, daf-18, dpy-10, edc-3, eel-1, egl-1, egl-19, fust-1, gyf-1, hsp-4, htz-1, hum-6, irg-1, laf-1, lin-5, mcm-5, mpk-1, mtl-1, nhr-6, nhr-8, npp-9, npp-10, pek-1, pqn-59, ptl-1, rab-7, rack-1, ruvb-1, sod-3, spt-16, F08G12.1, F13E6.1, H34C03.2, T13C2.6, T28D6.6, W08E12.7, Y38H6C.16, ZK1058.9
Cas9/sgRNA plasmid PCR
· Standard Q5 PCR protocol: 5 ng of pDD162 template, 61°C annealing, 30 cycles
· Optimized Q5 PCR protocol: 5 ng of pDD162 template, 65°C annealing, 15 cycles
Cas9/sgRNA plasmid KLD ligation
· KLD reaction: 1 µL of unpurified PCR product + 8.5 µL of KLD MM + 0.5 µL of KLD enzyme mix
· Standard KLD protocol: 5 min incubation at room temperature
· Optimized KLD protocol: 10 min incubation at room temperature, then 20 min incubation at 37°C
Purification of FP/SEC plasmid DNA fragments
Digested vector (e.g. pDD268 for mNeonGreen tags) and homology arm PCR products were purified using NEB® Monarch miniprep columns as follows:
(1) Dilute the volume of the digest and the PCRs to 40 µL each using ultrapure H2O.
(2) Add 200 µL of Qiagen buffer PB to each diluted reaction for a total of 240 µL.
(3) Purify the digestion and each homology arm separately using Monarch miniprep columns.
(4) Wash the columns with 400 µL of Monarch Wash Buffer 2.
(5) Elute the columns with 30 µL of Monarch Elution Buffer.
Quantification of FP/SEC plasmid DNA fragments
Purified DNA was quantified as follows:
(1) Run 1 µL of each purified DNA elution on the same 1% agarose gel containing ethidium bromide along with 2 µL of 0.02 µg/µL of DNA ladder standard.
(2) DNA band intensity was quantified relative to ladder using a Bio-Rad EZ Gel Doc system and Bio-Rad Image Lab 6.0 software.
(3) For each DNA fragment: [ng DNA x 1000]/[bp of DNA x 650 daltons] = # pmol/µL
Protocol variants for Gibson assembly of FP/SEC plasmid DNA fragments
· Previously published protocol (Dickinson et al. 2015): 4 µL of homology arms + 1 µL of digested vector + 5 µL of NEBuilder® HiFi DNA Assembly master mix
· NEBuilder® HiFi DNA Assembly protocol: 1:1 ratio of homology arms to digested vector, each fragment at 0.05 pmol in the reaction
· Glow Worms optimized protocol: 2:1 ratio of homology arms to digested vector, homology arm fragments at 0.004 pmol each and digested vector at 0.002 pmol each (i.e. 0.002 pmol of SEC fragment and 0.002 pmol of AmpR fragment).
FP/SEC plasmid Gibson assembly reaction
Gibson assembly reactions were incubated at 50°C for 1 hour and then either frozen at -20°C or used immediately for bacterial transformation.
Reagents
| Reagent | Supplier/Cat# or Recipe |
| T4 polynucleotide kinase | NEB® M0201S |
| T4 DNA ligase | NEB® M0202S |
| T4 DNA ligase buffer | NEB® B0202S |
| DpnI | NEB® R0176S |
| “Homemade” KLD master mix (MM) | 1 µL of T4 DNA ligase buffer + 9 µL of ultrapure H2O |
| “Homemade” KLD enzyme mix | 16.7 µL kinase + 1 µL ligase + 8.3 µL DpnI |
| Monarch miniprep kit | NEB® T1010L |
| PB buffer | Qiagen 19066 |
| DNA ladder standard | NEB® N3200S |
| HiFi DNA assembly master mix | NEBuilder® E2621L |
Acknowledgments
We would like to thank the 41 Glow Worms undergraduate students that took part in the design and creation of many of the plasmids analyzed in this study.
References
Funding
This work was supported by the Freshman Research Initiative of the Texas Institute for Discovery Education in Science of the College of Natural Sciences at The University of Texas at Austin, a fellowship award via The University of Texas at Austin Inventors Program (ED), and National Institute of Health grant R01 GM138443 (DJD). DJD is a CPRIT Scholar supported by the Cancer Prevention and Research Institute of Texas (RR170054).
Reviewed By
Jordan WardHistory
Received: October 7, 2021Revision received: November 11, 2021
Accepted: November 11, 2021
Published: November 19, 2021
Copyright
© 2021 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
DeMott, E; Dickinson, DJ; Doonan, R (2021). Highly improved cloning efficiency for plasmid-based CRISPR knock-in in C. elegans. microPublication Biology. 10.17912/micropub.biology.000499.Download: RIS BibTeX
