Abstract
Cyanobacteria, the only prokaryotes able of oxygenic photosynthesis are important primary producers that play a key role in the fields of agriculture, aquatic ecology and environmental protection. Their versatile metabolism makes them interesting candidates for various biotechnological applications. Recently, a great progress has been made in the field of their genetic manipulations by the development of CRISPR-based approaches. However, most of the available plasmids are rather difficult to manipulate, which renders their use challenging. In this study, we used the CcdB toxin as a selection marker to improve Cpf1-based plasmids designed for genome-editing in cyanobacteria. Our results demonstrate that this selection increased the rate of success of plasmid construction, and thus of genome editing.
Description
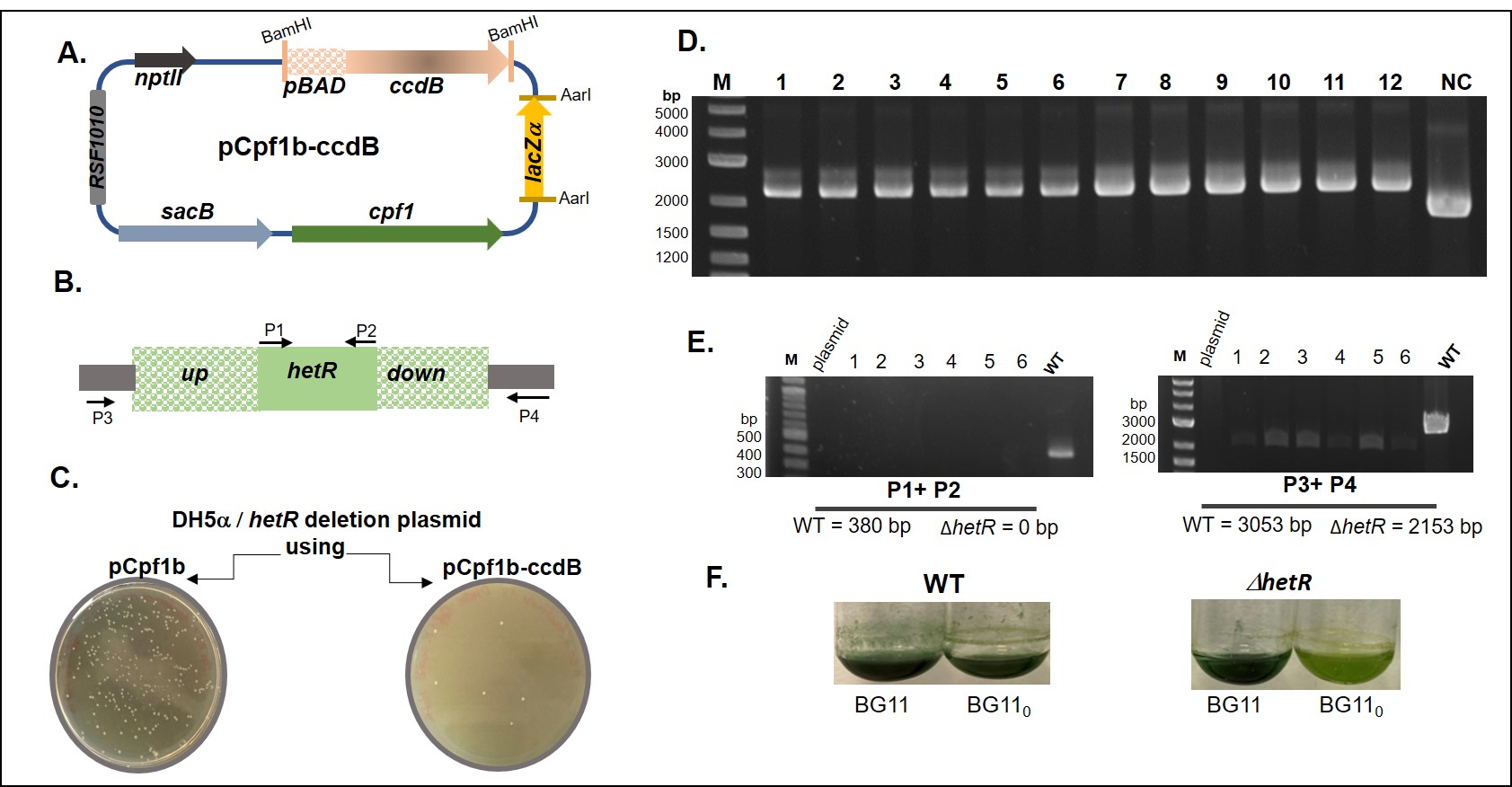
Recently, several CRISPR-based plasmids for editing cyanobacterial genomes have been developed. They derive mostly from vectors containing the low-copy number RSF1010 replication origin, and are of large size (> 10 kB). For instance, a CRISPR editing plasmid expressing the Cpf1 nuclease from Francisella novicida has been developed and shown to be efficient for editing cyanobacterial genomes (Ungerer and Pakrasi 2016). Later, this plasmid has been improved, notably by introducing the sacB counter-selection marker to optimize the loss of the editing plasmid once the genome-editing achieved (Niu et al. 2019). These plasmids are powerful tools, as their use has significantly decreased the time required to obtain mutants in cyanobacterial model strains. However, it has been largely reported that cloning in plasmids bearing the RSF1010 replication origin is difficult using techniques relying both on restriction enzymes, and approaches based on isothermal assembly. The copy number of RSF1010-type plasmids has been evaluated to be between 0.5 to 11.8 in Escherichia coli (Jahn et al. 2016), which explains the low yield of DNA obtained after plasmid extraction, and ineffective use in cloning procedures. In addition, the ability of RSF1010 plasmids to self-mobilize has also been suggested to explain their poor use as cloning-vectors (Taton et al. 2014). For all these reasons, we decided to first test our ability to manipulate the Cpf1-CRISPR plasmids by deleting the hetR gene of Nostoc PCC 7120 because the phenotype of the mutant is well defined (see below). In our hands, the cloning of the spacer was not a limiting-step due to the possibility of identification of positive clones by alpha-complementation (Ungerer and Pakrasi 2016). However, the insertion of the recombination platform (RP) –which contains the regions upstream and downstream to the gene to be deleted- was rather difficult, as a high number of negative clones are usually obtained (our success rate was less than 5%). In order to improve the cloning-success with this plasmid, we sought to introduce a screen that would allow direct selection of recombinant clones that have integrated the RP. To do this, we modified the plasmids pCpf1b (conferring resistance to kanamycin/neomycin) and pCpf1–sp (conferring resistance to spectinomycin) (Niu et al. 2019) by introducing the ccdB gene encoding a toxin that inhibits the DNA gyrase activity (Bernard and Couturier 1992). For this, the ccdB gene under the control of the pBAD promoter was amplified from the NM580 strain (Battesti et al. 2015) and cloned into the BamHI site of pCpf1b and pCpf1-sp plasmids. The XL1 Blue strain producing the anti-toxin CcdA was used for this experiment to inhibit the action of CcdB (Bernard and Couturier 1992). Over 100 colonies screened only 2 had the ccdB gene. A map of the obtained recombinant plasmid when pCpf1b was modified, is shown in Figure 1A. For the use of this plasmid for gene editing, the RP must be cloned between the BamHI sites, thus replacing the ccdB gene. This cloning must be performed in a strain that does not express the ccdA anti-toxin gene and arabinose must be added to the medium to induce expression of the toxin-encoding gene. Therefore, only clones that have integrated the RP are viable as the negative clones are counter-selected by the CcdB-induced lethality.
To analyze the efficiency of ccdB, we used the resulting pCpf1b–ccdB plasmid to delete the hetR gene of Nostoc PCC 7120. This cyanobacterium is able to fix dinitrogen thanks to the nitrogenase enzyme (Elhai and Wolk 1990). In this bacterium, nitrogen fixation relies on the ability to differentiate micro-oxic cells, that host the oxygen-sensitive nitrogenase (Flores and Herrero 2010). HetR is the master regulator of cell differentiation and is therefore essential when dinitrogen is the only nitrogen source available (Buikema and Haselkorn 2001). The scheme presented in Figure 1B describes the upstream and downstream regions used as the RP sequence to construct the hetR deletion mutant. The RP for hetR deletion was inserted in the pCpf1b-ccdB plasmid digested by BamHI (Figure 1A). For the selection of the E. coli recombinant clones, arabinose was added to the selective medium. The images of Figure 1C compare the total number of clones obtained when the cloning was achieved using the original (pCpf1b) or the modified (pCpf1b-ccdB) plasmids in a representative experiment. The results of Figure 1D show the analysis of recombinant clones chosen randomly during four independent experiments. The presence of the RP sequence was analyzed by PCR using the RP fw and rv primers and the pCpf1b–ccdB plasmid was used as a negative control. The expected amplicon size is 1734 pb for the pCpf1b–ccdB plasmid and 2186 pb in the case of successful RP cloning. As observed in Figure 1D, all the analyzed clones were positive which indicates that the ccdB gene is indeed an efficient selective marker. One of these plasmids was used to insert the hetR specific spacer. To optimize the targeting of the gene to be edited and its cleavage by Cpf1, we used the recently reported ChopChop software (Labun et al. 2019) to choose the spacer sequence (see Methods). The spacer sequence was cloned as explained in the Methods section. Positive colonies were identified by the alpha-complementation procedure. One of the resulting pCpf1-ccdB-ΔhetR plasmids was introduced by conjugation in Nostoc to delete the hetR gene. The PCR analysis of 6 clones obtained 15 days after the conjugation step demonstrated that all of them displayed the deletion (Figure 1E). As expected for a hetR mutant, the obtained strains were unable to sustain growth in the absence of combined nitrogen (Figure 1F). Additionally, we have used these improved plasmids to delete numerous other genes in a variety of on-going projects and a similar rate of success for the cloning of the RP sequences was obtained. We therefore conclude that, compared to their parental plasmids, the pCpf1-ccdB plasmids significantly optimize genome editing procedure in cyanobacteria.
Methods
Request a detailed protocolMethods
Cyanobacterial strains growth
Nostoc PCC 7120 and derivatives were grown in BG11 medium (Rippka R 1979) at 30 °C under continuous illumination (40 µE m-2s-1). When appropriate, media were supplemented with neomycin at a final concentration of (50 μg mL−1). Conjugation of Nostoc was performed as described in reference (Cai and Wolk 1990). To assess the phenotype of the hetR deletion mutant the strains were in BG110 (BG11 without sodium nitrate).
Construction of the ccdB-editing plasmid
The pBAD-ccdB sequence was amplified from the genome of the NM580 strain using the ccdB fw and ccdB rv primers. The In-Fusion technique (Takara) was used to insert the obtained amplicons in the pCpf1b and pCpf1-sp which were first linearized by BamH1. The XL1-Blue strain was used for transformation and the recombinant clones were analyzed by PCR, using the ccdB fw and the ccdB rv primers. Positive clones were further analyzed by sequencing.
Construction of the pCpf1-ccdB-DhetR plasmid
1kpb upstream and downstream of the hetR gene were amplified using the hetR up fw/ hetR up rv and hetR down fw/ hetR down rv primers, respectively. The two resulting amplicons were inserted in the BamHI site of the pCpf1-ccdB plasmid using the In-Fusion approach. Recombinants clones were obtained in the DH5α strain grown in the presence of arabinose at a final concentration of 0,1%. The recombinant plasmid obtained was analyzed by sequencing and named pCpf1-ccdB/RP
The spacer sequence was designed using the ChopChop software (Labun et al. 2019). The size of the primers was set to 22 nucleotides and the 5’-PAM sequence was designed TTN, as advised by Niu T et. al 2019 (Niu et al. 2019). The spacer-hetR fw and spacer-hetR rv primers were annealed at 95°C to obtain the hetR spacer, which was then inserted in the AarI site of the pCpf1b-ccdB/RP. The plasmids were introduced in the DH5alpha strain and recombinant clones were identified on selective medium containing Xgal at a final concentration of 80 μg/ml and isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 0,5mM. Positive clones were further analyzed by sequencing.
Reagents
Escherichia coli strains
| Strain name | Genotype | Source |
| NM580 | MG1655 lacI-T1T2-zeoR-pBRplacO-kan-pBAD-ccdB. Mini-l-Red::tetR | MA Lab |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F´ proAB lacIqZ∆M15 Tn10 (Tetr)] | Stratagene |
| DH5 |
F– endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG purB20 φ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK–mK+), λ– | Invitrogen |
Plasmids
| Name | Description | Source |
| pCfp1b | Cpf1-based CRISPR plasmid with kanamycin resistance marker and SacB counter selection marker | Addgene (#122187) |
| pCfp1-sp | Cpf1-based CRISPR plasmid with spectynomycin resistance marker and SacB counter selection marker | Addgene (#122186) |
Sequences of the primers used in this study
| Primer name | Sequence (from 5’ to 3’) | Experiment |
| ccdB fw | TAGATCTCATGGATCCAATTGTCTGATTCGTTACCAATT | Cloning of the pBAD-ccdB sequence in the pCpf1b and pCpf1-sp plasmids |
| ccdB rv | TTGCCATTGCGGATCCTTATATTCCCCAGAACATCAGGTT | |
| spacer hetR fw | agatAGCATAAGTTACCCAGCAATCT | Cloning of the hetR spacer |
| spacer hetR rv | agacAGATTGCTGGGTAACTTATGCT | |
| hetR up fw | ATATCTAGATCTCATGGATCCTGGTATTGGCAAAATACAAAATCCC | Cloning of the hetR upstream region: obtaining the RP |
| hetR up rv | CTCTGGGTGCATTACAAATAGTTGAATAGCACGC | |
| hetR down fw | TATTTGTAATGCACCCAGAGTGAATAAAAGTACT | Cloning of the hetR upstream region: obtaining the RP |
| hetR down rv | CGTTGTTGCCATTGCGGATCCTCGCGGATGATGGTATTAAGCT | |
| RP fw | CCTTTTGTATTAGTAGCCGG | Analysis of the insertion of the RP sequence |
| RP rv | TCTAGAGTCGAGCGCAA | |
| P1 | ATCTGATCAAGCGTCTTGG | Analysis of the deletion of hetR |
| P2 | TTCTTCACTTGTGAGGCTTG | |
| P3 | AAACATTGCCCAAATATTCG | |
| P4 | TTCTTCACTTGTGAGGCTTG |
Acknowledgments
The authors thank Dr Aurélia Battesti (Mireille Ansaldi (MA) Lab) for providing the NM580 strain.
References
Funding
This research was supported by the “Agence Nationale pour la Recherche Scientifique” (ANR-18-CE05-0029).
Reviewed By
AnonymousHistory
Received: November 17, 2021Accepted: January 10, 2022
Published: January 12, 2022
Copyright
© 2022 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Citation
Menestreau, M; Rachedi, R; Risoul, V; Foglino, M; Latifi, A (2022). The CcdB toxin is an efficient selective marker for CRISPR-plasmids developed for genome editing in cyanobacteria. microPublication Biology. 10.17912/micropub.biology.000512.Download: RIS BibTeX
